
Abstract
Tenofovir disoproxil fumarate (TDF), a prodrug of tenofovir, is a nucleotide reverse transcriptase inhibitor. It is administered orally at 300 mg once daily in combination with an additional nucleoside reverse transcriptase inhibitor (NRTI), typically emtricitabine, and an additional antiretroviral such as a non-nucleoside reverse transcriptase inhibitor (NNRTI), an integrase inhibitor or a protease inhibitor (PI). After hydrolysis in the lumen of the gastrointestinal tract, tenofovir is taken up by cells and subsequently phosphorylated to the active tenofovir-diphosphate (TFV-DP) moiety. TFV-DP is incorporated into viral DNA and terminates DNA elongation. Tenofovir is excreted by the kidneys through glomerular filtration and tubular secretion. Although few drug-drug interactions occur, tenofovir plasma concentrations may increase in the presence of other renally eliminated drugs. Safety and efficacy of TDF have been demonstrated in treatment-naïve and treatment-experienced patients. Although generally well tolerated, some studies suggest an increased risk for adverse events, such as bone toxicity and/or nephrotoxicity with long term TDF use. Patient adherence to TDF-containing regimens is improved over other antiretrovirals due to its once daily administration and low toxicity profile. Its use as pre-exposure prophylaxis against HIV is currently being investigated.
Introduction
Treatment of human immunodeficiency virus (HIV) has evolved over the past 30 years from monotherapy with the NRTI zidovudine (ZDV) to combination therapy including three or more antiretrovirals from at least two different classes. This combination therapy, referred to as highly active antiretroviral therapy or HAART, has reduced HIV-related morbidity and mortality.1,2 However, long-term therapy with HAART is often limited by adverse side effects and mutations in the genetic structure of HIV that confer resistance to antiretrovirals.3–6
The Department of Health and Human Services recommends various HAART regimens for the initial treatment of HIV and for those patients with treatment-resistant virus. 7 Options for initial therapy include a protease inhibitor (PI) based, non-nucleoside reverse transcriptase inhibitor (NNRTI) based, or an integrase inhibitor based regimen in combination with two NRTIs, commonly emtricitabine (FTC) and tenofovir disoproxil fumarate (TDF). TDF has been thoroughly studied and is generally well tolerated; thus, it is highly used in treatment naïve and experienced patients. 8 When used in combination at an oral dose of 300 mg once daily, TDF reduces HIV RNA in patients who are treatment-naïve, treatment-experienced or exhibiting resistant mutations. 9 Taken together, these characteristics make TDF an optimal choice for use as part of HAART. The following review summarizes our current understanding of the clinical pharmacology, efficacy, and safety of tenofovir based upon data collected from clinical trials in healthy volunteers and HIV-infected adults, children, and adolescents. The clinical use of TDF will be discussed as well as the future use of TDF in the role of pre-exposure prophylaxis (PrEP).
Pharmacology
Mechanism of action
Tenofovir is chemically identified as 9-
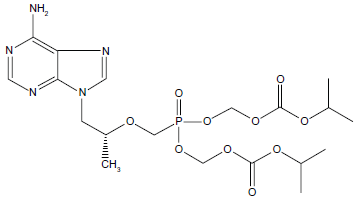
Chemical structure of tenofovir.
Pharmacokinetics
Absorption, bioavailability, and distribution
TDF is a water-soluble prodrug administered to yield the active intracellular TFV-DP. Addition of two esters increases the lipophilicity of the drug, thereby improving its permeability, biological stability, and oral bioavailability compared with tenofovir.
24
For example, tenofovir absorption in Caco-2 intestinal mucosal monolayers of mice and dogs increased 18% and 13%, respectively with TDF compared with tenofovir.13,14
Intravenous (IV) tenofovir dosed at 1 and 3 mg/kg to HIV-infected patients produced a volume of distribution (Vd) of 1.3 L/kg and 1.2 L/kg, respectively; mean steady-state Vd of TDF 300 mg QD was approximately 800 mL/kg.26,27 Furthermore, tenofovir is not highly protein bound to human plasma proteins (<1.0%) or to serum proteins (<7.2%) over a concentration range of 0.01-25 mg/L. Tenofovir penetration into various tissues and fluids after oral administration suggests it is able to enter extravascular compartments, such as the genital tract, by passive or facilitated diffusion and/or active transport due to its small size and degree of ionization. 28
Metabolism and elimination
Early
Pharmacokinetics in special populations
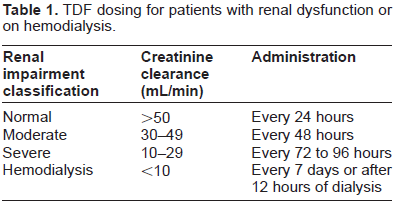
Pharmacokinetic data collected from studies conducted in healthy volunteers and HIV-infected patients demonstrate no effect of gender, age, or weight on tenofovir pharmacokinetics in adults receiving TDF. 33 Tenofovir is not metabolized by the liver, and liver dysfunction has demonstrated little to no impact on tenofovir pharmacokinetics. In contrast, elimination of tenofovir can be altered in patients with renal impairment. Mean ± standard deviation (SD) oral clearance (CL/F) of TDF decreased from 1,043.7 ± 115.4 mL/min in patients with normal renal function (CrCL: 50 mL/min) to 444.4 ± 209.8 mL/min in patients with moderate renal dysfunction (CrCL: 30-49 mL/min) and 177.0 ± 97.1 in patients with severe impairment (CrCL: 10 and 29 mL/min). 9 Tenofovir AUC0-∞ increased approximately 275% in patients with moderate dysfunction and 733% in patients with severe dysfunction compared to patients with normal renal function. These parameters were not significantly altered in patients with mild renal impairment. For those patients with end stage renal disease receiving hemodialysis, tenofovir was effectively filtered from serum with a median extraction coefficient of 54% and a four-hour session removing approximately 10% of the administered dose. 12 In summary, tenofovir pharmacokinetics are altered in patients with moderate or severe renal dysfunction and require dose adjustments as recommended in the dosage and administration section below and in Table 1.
TDF dosing for patients with renal dysfunction or on hemodialysis.
Children and adolescents
TDF once daily is also approved for the use in children and adolescents. Initial pediatric indication in patients ≥ 12 years of age and weighing at least 35 kilograms (kg) was gained based on steady-state pharmacokinetic data from only eight HIV-infected patients 12 to < 18 years of age receiving TDF 300 mg once daily. 9 Mean ± SD tenofovir Cmax and AUCτ were 0.38 ± 0.13 mg/L and 3.39 ± 1.22 mgxh/L, respectively. A separate study evaluated tenofovir pharmacokinetics after a single dose and at steady-state in 16 patients between six and 16 years of age. 27 The target TDF dose was 175 mg/m2 in anticipation of achieving TDF exposures similar to that achieved in adults after a 300 mg dose. The median TDF dose was 209 mg/m2, higher than the target dose due to restraints of dosing with a 75 mg investigational tablet. The geometric mean (GM) tenofovir AUC increased from 2.2 mgxh/L on day one to 2.9 mgxh/L at week four, exposures which are lower than the mean tenofovir AUC of 3.3 mgxh/L after a single dose and 3.4 mgxh/L at steady-state in adults. 25 Although the median TDF dose administered to children was higher than the target dose, the mean tenofovir AUC and Cmax were 34% and 27% lower, respectively, compared with adults. Renal clearance was approximately 1.5 fold higher in children and urinary recovery of 20% was similar between the two groups. Thus, the authors concluded that a higher renal clearance instead of lower oral bioavailability resulted in lower systemic exposure in children even at higher doses.
The investigational 75 mg tablet never became available for use in the clinical setting. Thus, limited pharmacokinetic data of TDF 300 mg in children and adolescents led a group of investigators to conduct an observational pharmacokinetic study of TDF-based regimens in 47 patients eight to 18 years of age. 34 Steady-state tenofovir pharmacokinetics were evaluated in combination with Food and Drug Administration (FDA)-approved doses of efavirenz (EFV), lopinavir/ritonavir (LPV/rtv)) or atazanavir/ritonavir (ATV/rtv). Median (range) serum creatinine and creatinine clearance at baseline were 0.6 (0.3 to 1.1) mg/dl and 154 (74.8 to 267.6) ml/min/1.73 m2, respectively, indicating no renal dysfunction within the patient population. GM (90% Confidence Interval (CI)) tenofovir AUC and Cmin in patients receiving EFV were 2.9 (2.5-3.4) mgxh/L and 0.07 (0.05-0.09) mg/L, respectively. In patients receiving LPV/rtv, tenofovir AUC and Cmin were 3.0 (2.5-3.6) mgxh/L and 0.06 (0.05-0.08) mg/L, respectively. In patients receiving ATV/rtv, tenofovir AUC and Cmin were 3.6 (3.1-4.2) mgxh/L and 0.07 (0.06-0.09) mg/L, respectively. Informal comparisons of TDF pharmacokinetics across the three groups indicated no differences in tenofovir exposure. These results were not expected since higher tenofovir exposure has been noted in adults concomitantly receiving LPV/rtv or ATV/rtv (as discussed in the section Drug Interactions/Protease Inhibitors). In summary, the investigators supported the use of TDF 300 mg once daily in patients eight to 18 years of age. Most recently, the FDA approved three lower-strength once-daily tablets of TDF in doses of 150 mg, 200 mg and 250 mg for ages six to 12 years. 9 The recommended dose in this patient population is 8 mg/kg/day. An oral powder formulation for children ages two to five years was also approved.
Drug Interactions
TDF is used in combination with other antiretrovirals to treat HIV infection and often, HIV-infected patients receive concomitant medications to treat other disease states. Thus, the pharmacokinetics of TDF in combination with other antiretrovirals and non-antiretrovirals have been extensively evaluated. As previously mentioned, tenofovir does not inhibit or induce
Nucleoside reverse transcriptase inhibitors (NRTIs)
NRTIs are considered the backbone of antiretroviral therapy and are commonly used in combination with at least one additional NRTI. These antiretrovirals interrupt HIV replication by mimicking endogenous nucleosides and terminating proviral DNA chainformation. 35 ABC is a guanosine analogue, ddI an adenosine analogue, 3TC and FTC are cytidine analogues, and ZDV and d4T are thymidine analogues. All of the nucleoside analogues require triple phosphorylation to the active triphosphate moiety. TDF, however, only requires double phosphorylation to the active TFV-DP and is considered a nucleotide analogue. Regardless of the number of phosphorylation steps required, competition amongst the analogues for phosphorylation by cellular kinases can result in drug-drug interactions. Additionally, all NRTIs are renally eliminated except for ABC which is metabolized by alcohol dehydrogenase and glucuronyl transferase. Competition between tenofovir and other antiretrovirals for drug transporters responsible for renal elimination further increases the potential for interactions. No interactions have been noted between TDF and 3TC or FTC; therefore, they are commonly administered together. 36
Abacavir (ABC)
Several large, multicenter studies reported surprisingly suboptimal virologic results in patients receiving once or twice daily ABC-containing HAART regimens, including those with TDF.37,38 As a result, the pharmacologic properties of these agents in combination were evaluated. One study conducted in eight HIV-infected patients receiving a single dose of ABC 300 mg twice daily (BID) with and without TDF 300 mg once daily (QD) report a geometric mean ratio (GMR) of 1.10 for ABC AUC0-∞ and 1.12 for ABC Cmax in the presence of TDF. In the presence of ABC, the GMR for tenofovir AUC and Cmax were 1.04 and 0.92, respectively, compared to historical controls receiving TDF alone. 39 These results did not suggest a pharmacokinetic interaction between the two agents. A second study evaluated the intracellular antiretroviral concentrations in 15 HIV-infected patients taking TDF 300 mg QD, ABC 300 mg BID, and a third NRTI who were switched off TDF or ABC for an NNRTI or PI. 40 Intracellular nucleotide concentrations of the continued drugs were not altered when TDF or ABC were switched off, suggesting that a pharmacody-namic drug-drug interaction on an intracellular level did not occur. Although a pharmacokinetic/pharmacodynamic drug-drug interaction does not explain the decreased efficacy of TDF and ABC, these drugs are not recommended as a preferred regimen in combination as the entire NRTI backbone or as part of a triple NRTI regimen. 7
Didanosine (ddI)
Initial evaluations of TDF in combination with ddI as either the buffered or enteric-coated formulation demonstrated a 28% increase in ddI Cmax and a 40% increase in ddI AUC. 41 The intracellular exposure of ddI also increased and is thought to be a result of TDF inhibiting the enzyme purine nucleoside phosphorylase (PNP), which phosphorolysis ddI. 42 An initial decrease in HIV RNA was noted when using these drugs in combination. However, a 12 week study reported virologic failure in 91% of patients receiving TDF, ddI and 3TC. 43 Adverse events such as pancreatitis, symptomatic hyperlactatemia, lactic acidosis and severe CD4 lymphopenia with prolonged use have been frequently reported. As a result, co-administration of TDF and ddI is not generally recommended. 7
Non-nucleoside reverse transcriptase inhibitors (NNRTI)
NNRTIs noncompetitively bind to the p66 subunit of viral reverse transcriptase and induce a conformational change in the enzyme that alters the active site and limits its viral activity. 44 EFV, NVP, etravirine (ETR) and rilpivirine (RPV) are the four NNRTIs currently approved for use in the treatment of HIV and are commonly combined with two NRTIs. As a class, NNRTIs are metabolized by various isoenzymes of CYP and can be potent inducers or inhibitors of the enzyme family. Therefore, interactions between TDF and NNRTIs are not expected. For example, administration of TDF and ETR to healthy volunteers resulted in a 19%, 18%, and 26% decrease in ETR Cmax, Cmin, and AUC12 and a 15% increase in tenofovir Cmax and AUC. 45 All changes were considered clinically insignificant. NVP trough concentrations with and without TDF were 3,420 ng/mL and 3,260 ng/mL, respectively, suggesting no impact of TDF on NVP exposure. 46 Co-administration of RPV and TDF to healthy volunteers for eight days revealed no changes in RPV pharmacokinetics while tenofovir AUC, Cmax, and Cmin increased approximately 20%. 47 Similar results have been noted for TDF in combination with EFV. 48 The absence of a clinically significant pharmacokinetic interaction combined with the potency of these two antiretrovirals has led to their increased use in combination (as discussed in the Adherence section).
Protease inhibitors (PI)
PIs bind directly to HIV protease enzymes preventing cleavage of polypeptides into smaller proteins which halts maturation and the ability of the virus to infect new cells. 49 There are currently eight PIs approved for use in the treatment of HIV. However, this discussion is limited to those PIs used most frequently in combination with TDF and resulted in increased plasma concentrations of tenofovir. 50 Of note, all PIs listed below discuss the use of low-dose ritonavir (RTV) in combination with the PI. RTV is a potent inhibitor of CYP3A4 and p-glycoprotein (Pgp) drug transporters.50,51 Combined use of low dose RTV with other PIs increases the bioavailability and plasma concentrations of the concomitantly administered PI.
Lopinavir/ritonavir (LPV/rtv)
Multiple studies have evaluated LPV/rtv in combination with TDF. One study reports a decrease in LPV AUC and Cmax by approximately 15% in the presence of TDF. 52 In contrast, a cross-over study conducted in 23 healthy volunteers reports similar LPV exposure with and without TDF 300 mg QD.53 Tenofovir AUC, Cmax and Cmin increased 32%, 15% and 51%, respectively in the presence of LPV/rtv 400/100 mg. Co-administration was generally safe, well tolerated and associated with infrequent evidence of nephrotoxicity, supporting clinical use of these antiretrovirals in combination. A separate study reported 15% slower tenofovir renal clearance in HIV-infected patients taking LPV/rtv (n = 15) compared with those patients not taking a PI (n = 15). 54 A drug-drug interaction involving MDRP 1 and 2 transporters in the kidneys likely accounts for the reported decreased in tenofovir renal clearance. However, more extensive studies are necessary to completely understand the mechanism and risk of potential nephrotoxicity. Consistent monitoring of kidney function is encouraged when using LPV/r and TDF in combination.55,56
Atazanavir/ritonavir (ATV/rtv)
The recommended dose for ATV in HIV-infected patients is 400 mg once daily. 26 Initial pharmacokinetic data demonstrated a significant decrease in mean ATV AUC of 25% and Cmin of 40%, respectively, when combined with TDF compared with ATV alone. 57 As a result, when co-administered with TDF 300 mg QD, ATV 300 mg should be used in combination with RTV 100 mg. A study performed in 10 HIV-infected patients evaluated the pharmacokinetics of RTV-boosted ATV with and without TDF. 58 In the presence of TDF, there was a trend towards lower ATV and RTV concentrations but the 25% decrease in ATV AUC was the only significantly reduced pharmacokinetic parameter. In contrast, a separate study evaluating the same combination in healthy volunteers suggested ATV AUC only decreased by 11%. 59 Tenofovir AUC and Cmin increased 38% and 33%, respectively.
Darunavir/ritonavir (DRV/rtv)
Increased tenofovir plasma concentrations have also been reported when co-administered with DRV/rtv. 60 Data were collected from 12 HIV-negative volunteers participating in a crossover study of DRV/rtv 300/100 mg BID with and without TDF 300 mg QD. In the presence of DRV/rtv, tenofovir Cmin, Cmax and AUC24h increased 37%, 24% and 22%, respectively. DRV Cmin, Cmax and AUC increased 12%, 24% and 21%, respectively in the presence of TDF. Urinary excretion of unchanged DRV or tenofovir was not significantly altered. This study evaluated a standard TDF dose in combination with a DRV/rtv dose lower than the approved dose of 600/100 mg BID. However, results from this study were considered applicable to the standard dose since a 50% increase in DRV dose in the presence of RTV 100 mg increases DRV exposure by only 29%. 61 Taken together, these data suggest using standard doses of TDF and DRV/rtv in combination.
In summary, tenofovir concentrations increase in the presence of several RTV-boosted PIs; however the mechanism of this interaction is unknown. Since tenofovir is not a substrate for CYP enzymes, a drug-drug interaction altering metabolism is unlikely. Several investigators have theorized that increased tenofovir concentrations result from a drug-drug interaction occurring on a molecular level in the kidneys. RTV is a potent inhibitor of Pgp and MDRP-2 drug transporters.
62
Inhibition of efflux transport by RTV could theoretically increase tubular concentrations of tenofovir in the kidneys. This hypothesis, however, remains unproven. Others theorize that tenofovir transport into the renal proximal tubule cells by organic anion transporters may be inhibited. However, data suggest that tenofovir plasma concentrations would occupy only a limited fraction of the renal transport capacity and PIs have a low potential to interfere with active tubular secretion of tenofovir.
63
Interestingly, several studies report a decline in renal function in patients receiving antiretroviral therapy with TDF and RTV-boosted PIs. A prospective observational cohort study evaluated 432 antiretroviral naïve patients receiving a NRTI based regimen. Estimated glomerular filtration Rate (eGFR) was compared between patients receiving TDF plus RTV-boosted PI and TDF plus an NNRTI. Patients receiving TDF co-administrated with RTV-boosted PI had a greater decline in eGFR than those taking TDF with an NNRTI at 6 months (
Chemokine receptor antagonists
Currently, one chemokine receptor antagonist is approved for use in the treatment of HIV. Maraviroc reversibly binds the CCR5 co-receptor, blocking the V3 loop interaction and inhibiting fusion of the cellular membranes to reduce insertion of the virus into the host cell. 66 In a crossover clinical trial that enrolled 11 healthy male volunteers, TDF was co-administrated with maraviroc and showed no significance changes in AUC or Cmax for either drug. 67
Integrase inhibitors
Integrase inhibitors are the newest class of antiretrovirals. Raltegravir (RAL) is the only agent approved in this class. The drug competitively inhibits strand transfer reaction by binding metallic ions in the active site and restricting proviral DNA covalently linked to cellular DNA. 68 Co-administration of TDF 300 mg and RAL 400 mg BID was evaluated in an open-label, three period study in 10 healthy subjects. RAL AUC increased 49%, Cmax increased 64% and Cmin remained unchained in the presence of TDF. Increased RAL exposure was considered insignificant due to the lack of safety issues reported with maximum RAL plasma concentrations across several studies. In contrast, TDF showed a decrease in AUC and Cmin by 10% and 13%, respectively, but these changes were not considered clinically significant. In conclusion, dosage adjustments are not needed when TDF and RAL are used in combination. 69
Non-antiretroviral drugs
HIV-infected patients often present with other disease states that require medications for treatment. Overall, few drug-drug interactions occur with TDF but instead interactions occur with the other antiretrovirals co-administered with TDF in combination therapy. Acyclovir, cidofovir, foscarnet, ganciclovir and amphotericin B should be avoided in combination with TDF. Although not extensively studied, these drugs are renally eliminated and may compete with TDF for renal tubular secretion, subsequently increasing tenofovir plasma concentrations. Table 2 summarizes changes in tenofovir pharmacokinetic parameters when TDF is co-administered with common non-antiretroviral drugs.
Pharmacokinetics of TDF and commonly administered non-antiretroviral medications.
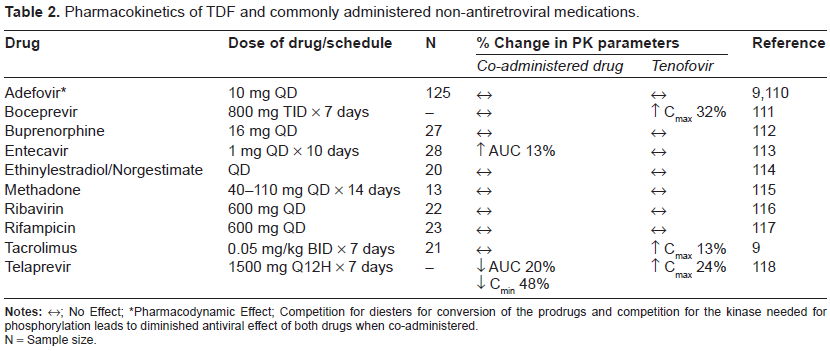
Pharmacodynamic Effect; Competition for diesters for conversion of the prodrugs and competition for the kinase needed for phosphorylation leads to diminished antiviral effect of both drugs when co-administered.
N = Sample size.
Resistance
TDF became available for clinical use during a time when clinicians were searching for second and third line therapies for treatment-experienced patients. Initial analyses of tenofovir
These findings were supported
Clinical Studies
Tenofovir was initially studied after a single IV dose and repeated for seven days at two dose levels (1.0 and 3.0 mg/kg) in 20 HIV-infected patients with a CD4 cell count ≥ 200 cells/mm3 and an HIV RNA ≥ 10,000 copies/mL. 76 Patients were randomly assigned in a 4:1 ratio to receive IV tenofovir 1 mg/kg (n = 8), IV tenofovir 3 mg/kg (n = 8), or placebo (n = 4). Study drug was administered once on day one followed by a seven-day washout period and then once a day as an IV infusion for seven days. On day 14, plasma HIV RNA decreased 1.1 log10 and 0.6 log10 from baseline for the 3 mg/kg and 1 mg/kg doses, respectively while HIV RNA increased 0.1 log10 from baseline for the placebo group. Systemic exposure of tenofovir increased significantly after 14 days of dosing at 3.0 mg/kg and was likely a result of the prolonged TFV-DP half-life.
Phase I/II studies of TDF began in 1997 when HIV-1 infected patients were randomized into a dose-finding study evaluating TDF 75, 150, 300 and 600 mg once daily versus placebo.
25
After 28 days of monotherapy, the median decrease in log10 HIV RNA was significantly greater at -1.22 log10 copies/mL for the 300 mg dose compared with -0.33 and -0.44 log10 copies/mL for the 75 and 150 mg doses, respectively. Although pharmacokinetic data demonstrated higher systemic exposure with 600 mg doses, decreases in HIV RNA were not significant compared with 300 mg doses suggesting that 300 mg achieves maximal anti-retroviral activity. A follow-up phase II study investigated placebo or multiple doses of TDF 75, 150 and 300 mg in combination with antiretroviral therapy in 186 treatment-experienced, HIV-1 infected patients experiencing incomplete virologic suppression.
77
At baseline, mean plasma HIV-1 RNA was 3.7 log10 copies/mL and CD4 count was 374 x 10
6
cells/µl. Patients had a mean 55 months of prior antiretroviral therapy and 94% had a nucleoside-associated resistance mutation. The average change from baseline log10 HIV RNA at 24 weeks was greatest for TDF 300 mg (-0.58 log10 copies/mL) and remained durable out to 48 weeks (-0.62 log10 copies/mL). However, median CD4 cell count did not change significantly at 24 weeks compared with baseline. A similar study design was utilized in study GS907 when 552 treatment-experienced patients with plasma HIV RNA between 400 and 10,000 copies/mL were randomized to receive TDF 300 mg or placebo in addition to their current antiretroviral therapy.
74
After 24 weeks of therapy, HIV-1 RNA decreased significantly in the TDF group compared with placebo (-0.61 log10 copies/ml vs. -0.03 log10 copies/ml,
One of the first comparative trials evaluated TDF 300 mg QD (n = 299) versus d4T 400 mg BID (n = 301), both in combination with 3TC 150 mg BID and EFV 600 mg QD in 602 treatment-naïve patients.
78
In an intent to treat (ITT) analysis, where missing or antiretroviral switch equaled failure, 80% of patients in the TDF arm achieved an HIV RNA level less than 400 copies/mL at 48 weeks compared with 84% of patients in the d4T group. Since the proportion of patients achieving an HIV RNA < 400 copies/mL at 48 weeks was similar between the two arms, TDF slightly missed the predefined criteria (-10% limit) for equivalence to d4T; however, it demonstrated non-inferiority to d4T at weeks 96 and 144. CD4 count was 263 cells/µl in the TDF group through week 144 and was comparable with the d4T group. A non-inferiority study compared TDF, FTC plus EFV QD with ZDV and 3TC BID plus EFV QD in 509 HIV-infected patients.
79
Data from this study suggested that a significantly greater response was seen in those patients receiving TDF-based regimens. For example, 81% of patients in the TDF containing arm maintained an HIV RNA less than 400 copies/mL compared with 77% in the ZDV/3TC arm (95% CI for the difference, 3% to 18%;
Safety and Tolerability
Clinical trials report that TDF is generally well tolerated compared with placebo in patients who are treatment naïve and treatment experienced. In treatment-naïve patients receiving a TDF-based or a d4T-based regimen, the incidence of Grade 3 or 4 adverse events (27% for TDF vs. 25% for d4T) or laboratory abnormalities (36% vs. TDF vs. 42% for d4T) after 144 weeks of treatment were not significantly different between arms.9,78 Less than 25% of all patients reported grade 2-4 adverse events with greatest incidence of headache (14% TDF vs. 17% d4T), pain (13% TDF vs. 12% d4T), abdominal pain (7% TDF and 12% d4T), diarrhea (11% TDF vs. 13% d4T), depression (11% TDF vs. 10% d4T) and rash events (18% TDF vs. 12% d4T). A similar incidence of adverse events was noted in treatment-naïve patients receiving TDF, 3TC and EFV compared with those receiving ZDV, 3TC and EFV. 80 Pooled data analysis from placebo-controlled studies in treatment-experienced patients state that 3% of patients discontinued usage of TDF due to adverse events during the initial 24 weeks and increased to only 9% over the mean treatment period of 95 weeks. 81 Gastrointestinal issues were the most prominent adverse event.
Significant differences in laboratory parameters were not noted after 24 and 48 weeks administration of TDF compared with placebo or when a TDF containing regimen was compared with a ZDV containing regimen.74,80 With the exception of higher fasting cholesterol (40% vs. 19%) and triglycerides (9% vs. 1%) in the d4T containing regimen, laboratory abnormities occurred at a similar frequency in the TDF containing regimen. Occasional elevations in serum creatinine and phosphorus levels were reported in clinical trials of treatment experienced patients taking TDF, but the levels did not warrant discontinuation of TDF. 81 Although long term studies comparing TDF with other antiretrovirals report similar adverse events, the potential for toxicity especially mitochondrial, bone and/or renal exists with long term TDF usage.
Mitochondrial toxicity
The risk of mitochondrial toxicity associated with TDF use appears similar to placebo and lower than d4T. Neuropathy was reported in 3% of patients receiving TDF compared with 10% of patients receiving d4T. 77 The incidence of lipodystrophy (3% vs. 19%) and lactic acidosis (0 vs. 3 occurrences) were also significantly lower for TDF-containing regimens. Pancreatitis was not reported in either arm. Cases of lactic acidosis and pancreatitis have been reported with clinical use of TDF, often in combination with ddI and the relationship between TDF and these toxicities have been difficult to establish.82–85
Nephrotoxicity
Animal studies report dose-limiting nephrotoxicity associated with TDF use. 86 This adverse event, however, was not observed in large, short-term clinical trials where renal safety profiles were similar between patients receiving TDF and placebo or other antiretrovirals.25,74 In contrast, 19 cases of TDF-associated proximal renal tubular dysfunction were reported in treatment-experienced patients receiving TDF therapy. 87 Renal dysfunction was noted, on average, 7 months after initiation of therapy and resolved approximately 5 weeks after discontinuing therapy. Subsequently, long-term renal safety was evaluated in large clinical trials. A long-term analysis evaluated renal safety in patients receiving TDF compared with d4T. 88 At week 144, mean serum creatinine did not change in the TDF group compared with a 0.1 mg/dl decrease from baseline in the d4T group. No patient experienced grade 4 (< 1.0 mg/dl) hypophosphatemia and no patient developed Fanconi's syndrome or proximal renal tubular dysfunction. A meta-analysis of 17 studies demonstrated a significantly greater loss of kidney function among patients receiving TDF compared with those receiving antiretroviral therapy without TDF (mean difference in calculated CrCL, 3.92 mL/min vs. 2.13-5.70 mL/min). 89 The evidence did not suggest that TDF leads to an increased risk of severe proteinuria, hypophosphatemia, or fractures. Thus, the authors support TDF use with regular monitoring of renal function in patients with normal or impaired kidney function.
Bone toxicity
Initial animal studies showed changes in bone mineral density (BMD) at tenofovir exposures 6 to 12 times higher than those observed in humans. No statistically significant changes in BMD over a dose range of 0-300 mg/day for 48 weeks were reported in 62 HIV-infected patients. 90 However, BMD at 144 weeks in a separate study decreased in TDF-treated patients significantly (2.2%) in the lumbar spine but not significantly at the hip (2.8%) compared with d4T-treated patients. 77 Subsequent studies have also suggested greater loss of BMD with TDF use. 91
Studies conducted in infant monkeys report stunted growth and bone toxicity, raising concerns about its use in children. 86 A phase I/II clinical trial of TDF 75 mg once daily in 18 HIV-infected children and adolescents reported a decrease in BMD of approximately 6% with a greater decrease seen in patients with a median Tanner Stage of one and an average age of 10.2 years. 92 Absolute lumbar spine BMD has also been reported to decrease in adolescents after 48 weeks of TDF treatment. 93 A separate study evaluating TDF 300 mg in six adolescents reported a decrease in BMD that correlated with higher drug exposure, specifically tenofovir AUC. 94 The authors suggested that higher tenofovir exposure was attributed to co-administration with RTV and the increased TDF dosage. A correlation was also noted between higher drug exposure and better virological response, as verified by a decrease in plasma HIV RNA. In another study, BMD did not decrease and virological suppression was maintained with TDF use in older children and adolescents. 95 Study results may have been skewed by the fact that these patients were previously treated with a d4T-based regimen, older in age than earlier studies, not receiving RTV, and receiving lower doses of TDF.
Patient Preference
Dosage and administration
The recommended dosage of TDF is 300 mg QD, without regard to meals, in combination with other anti-retroviral agents for the treatment of HIV-1 infection. 9 As previously mentioned, TDF is renally excreted, so dosage recommendations differ for patients with renal dysfunction. These recommendations, including dosing for patients receiving hemodialysis are listed in Table 1.9,96 TDF pharmacokinetics are not substantially altered in patients with hepatic impairment, thus dosage adjustments in this population are not necessary. TDF dosing in children and adolescents is based on age and weight. 9 Recent approval of smaller dosage formulations will likely increase TDF use in the younger population. Dosing recommendations are not available for patients aged >65 years. Since the elderly generally have significant overlapping comorbidities, including reduced drug clearance mechanisms, reduced cardiac function and other diseases requiring multiple concomitant medications, monitoring TDF adverse events is suggested in this patient population. 96 Urinalysis should be performed every six months in these patients. 7
Adherence
Adherence to antiretroviral therapy is strongly associated with successful virological and clinical outcomes and data suggest high rates of adherence to TDF-containing regimens.7,97,98 Once daily administration of TDF without regards to meals reduces pill burden and allows for an easier integration into daily life compared with drugs that must be taken with food or on an empty stomach. Furthermore, minimal toxicities and few drug-drug interactions reported with TDF increase patients’ adherence and affords this antiretroviral as a preferred drug amongst clinicians and patients. To further improve adherence, pharmaceutical companies have co-formulated commonly administered antiretrovirals into one dosage formulation. The first fixed dose combination (FDC) tablet approved by the FDA was Truvada®, which contains TDF 300 mg and FTC 200 mg to be administered QD. 99 Atripla®, which contains TDF 300 mg, FTC 200 mg and EFV 600 mg, was subsequently approved for QD dosing followed by Complera®, which contains TDF 300 mg, FTC 200 mg and RPV 25 mg for QD dosing.44,100,101
Place in Therapy
Treatment of HIV-infection
Recommendations for combination antiretroviral therapy in non-pregnant adults generally consist of two NRTIs and one drug from the following classes: NNRTI, PI (generally boosted with RTV), integrase inhibitor or a CCR5 antagonist. 7 According to these guidelines, preferred regimens are those antiretrovirals when used in combination, in large clinical studies, produced optimal and durable efficacy, favorable tolerability and toxicity profile, and ease of use. Among the four preferred regimens for treatment-naïve patients, TDF in combination with FTC are the recommended NRTIs in all four regimens.
Use during pregnancy and prevention of mother to child transmission
At the time of FDA approval, TDF use in humans during pregnancy had not been adequately studied. 9 Initial evaluations were conducted in monkeys receiving a daily dose approximately twice that administered to humans and resulted in decreased fetal growth and reduced fetal bone porosity within two months of starting maternal therapy. 102 However, no fetal structural abnormalities were noted prompting the FDA to assign TDF to a pregnancy Category B designation. Subsequently, guidelines recommended TDF use only in special circumstances, such as cases when intolerance or resistance prevent the use of other less toxic antiretrovirals or in women who have comorbidities or require concomitant medications that may limit drug choice. 103 Data released in 2011 by the Antiretroviral Pregnancy Registry showed no increase in overall birth defects in 1,092 pregnancies with first trimester exposure to TDF compared with the general population. 103 Pharmacokinetic evaluations of TDF during pregnancy showed similar exposure after a 300 mg dose compared with post-partum.104,105 As a result, guidelines were updated to recommend TDF as an alternative NRTI choice during pregnancy and a preferred NRTI in combination with 3TC or FTC in pregnant women with chronic hepatitis B co-infection.
Post-exposure prophylaxis (PEP) and PreExposure prophylaxis (PreP)
PEP involves the use of antiretroviral therapy by someone potentially exposed to HIV, either inside or outside the workspace. In these cases, the HIV statuses of all individuals are typically known. According to the United States Center for Disease Control, PEP treatment consists of a four-week program of two or three antiretrovirals. 106 Antiretrovirals chosen are based on the potential route of exposure to HIV, other drugs that are likely to interact with antiretrovirals, and the physical health of the person exposed to the virus. TDF is a commonly used NRTI in PEP programs because of its potency, tolerability, and dosing.
PrEP is a medical intervention applied before exposure to a disease to create defense mechanisms against the disease. In the case of HIV, this technique consists of administering antiretrovirals to HIV negative individuals to reduce the risk of contracting the virus. Ideally, stopping viral replication prior to viral DNA integration into the host cell's DNA by NRTIs or NNRTIs should prevent HIV transmission. Tenofovir is active in proliferating and resting cells allowing for sustained antiviral potency over a long duration.
Conclusion
TDF, the prodrug of tenofovir, is a NRTI approved at a dose of 300 mg QD to be administered in combination with other antiretrovirals for the treatment of HIV. After oral administration, TDF is hydrolyzed to tenofovir and subsequently phosphorylated twice inside the cell to the active TFV-DP moiety. Tenofovir is not metabolized by liver enzymes but is renally excreted as unchanged drug. Thus, drug-drug interactions with many co-administered drugs that are substrates, inhibitors and/or inducers of liver enzymes are avoided. However, caution should be taken when administering TDF with nephrotoxic drugs. Furthermore, patients with renal dysfunction require TDF dose adjustments. Tenofovir is a potent inhibitor of HIV replication, both
Author Contributions
Wrote the first draft of the manuscript: AMJ and JRK; Agree with manuscript results and conclusions: AMJ, IO, AS, EPA, JRK; Made critical revisions and approved final version: EPA, IO, AS, JRK. All authors contributed to writing and approved of the final manuscript.
Funding
1K23A10074390-01 from the National Institute of Allergy and Infectious Diseases (JRK).
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest. Provenance: the authors were invited to submit this paper.