
Abstract
Sitafloxacin (DU-6859a) is a new-generation oral fluoroquinolone with in vitro activity against a broad range of Gram-positive and -negative bacteria, including anaerobic bacteria, as well as against atypical bacterial pathogens. Particularly in Japan this antibiotic was approved in 2008 for treatment of a number of bacterial infections caused by Gram-positive cocci and Gram-negative cocci and rods, including anaerobia atypical bacterial pathogens. As compared to oral levofloxacin sitafloxacin was non-inferior in the treatment of community-acquired pneumonia and non-inferior in the treatment of complicated urinary tract infections, according to the results of randomized, double-blind, multicentre, non-inferiority trials. Non-comparative studies demonstrated the efficacy of oral sitafloxacin in otorhinolaryngological infections, urethritis in men, cervicitis in women and odontogenic infections. Most common adverse reactions were gastrointestinal disorders and laboratory abnormalities in patients receiving oral sitafloxacin; diarrhea and liver enzyme elevations were among the common. In the Japanese population sitafloxacin covers broad spectrum of bacteria as compared to carbapenems, whereas in the Caucasians its use is currently limited due to the potential for ultraviolet A phototoxicity. Sitafloxacin is a promising therapeutic agent which merits further investigation in randomized clinical trials of elderly patients.
Introduction
Infectious diseases account for one third of all deaths in people 65 years and older. Bacterial infections are common in elderly patients associated with morbidity and mortality in this population group. Age-related predisposing factors determine the decrease in the ability of the human body to prevent penetration of pathogenic microorganisms and to resist the development of infectious process. General and local factors contribute to the development of frequent infections in the elderly population.1,2 Among the general factors are to mention: dehydration, reduction of the protective barrier of the skin and mucous as a result of atrophic processes, immunodeficiency (notably reduced IgM activity and function of T-lymphocytes), micro-circulation disturbance, impaired consciousness, and poor personal hygiene.2,3 Urinary tract infections are the most common cause of bacteremia in older adults. Asymptomatic bacteriuria occurs frequently in the elderly; however, antibiotic treatment does not appear to be efficacious. 4
There are higher proportions of antibiotic resistance among bacteria in elderly patients, especially methicillin resistance among Staphylococcus aureus isolates across all specimen types, and resistance to several empiric antibiotics among Enterobacte-riaceae isolates in urine cultures.4,5 Older patients are at risk for COPD (chronic obstructive pulmonary disease) and emphysema, chronic bronchitis, and bronchiectasis. 6 Besides viral infections, bacterial infections play a role in acute exacerbations of COPD (AECOPD). Elderly patients are at risk for resistant bacterial organisms during their episodes of AECOPD. Organisms include the more-common bacteria implicated in AECOPD such as Haemophilus influenzae, Moraxella catarrhalis, and Streptococcus pneumoniae. Pseudomonas aeruginosa, Staphylococcus aureus, and Mycobacteria other than tuberculosis (MOTT) are more often seen in AECOPD episodes. 6 Appropriate therapy or newer antimicrobial compounds are required in such episodes while majority of pathogens are becoming for more resistant to the current available compounds. Since the last decades of the 20th century serious infections caused by bacteria that have become resistant to commonly used antibiotics have become a major global healthcare problem.7,8 Antibiotic resistance, initially a problem of the hospital setting associated with an increased number of hospital-associated infections usually in critically ill and immunosuppressed patients, has now extended into the community causing severe infections difficult to diagnose and treat. The molecular mechanisms by which bacteria have become resistant to antibiotics are diverse and complex. Bacteria have developed resistance to all different classes of antibiotics discovered to date. 8 Fluoroquinolones or quinolones remain as important antibiotics administered in bacterial infections.9,10 Quinolones comprise a relatively large, growing and most interesting group of antibiotics which have made a major impact on the field of antimicrobial chemotherapy, particularly in the past decade. 11 The first quinolone to be administered for patient treatment was nalidixic acid, discovered by Lesher and co-workers in the 1960s, which was generated from the antimalarial agent chloroquine.12–14 It was active against some Gram-negative bacteria and had limited clinical efficacy because of its high protein binding (approximately 90%) and short half life (about 1.5 h). 15 Unfortunately, bacteria rapidly develop resistance to this agent and other quinolones.16–20
Quinolones have been widely administered since the introduction norfloxacin which was patented in 1978. By modification of nalidixic acid structure the synthesis of norfloxacin, as the first fluoroquinolone, was enabled.11,21 This antibiotic exhibited enhanced activity for Gram-negative bacteria, including Pseudomonas aeruginosa.11,21 Despite some rather unpromising features of the early compounds, persistent efforts have been made over the years to produce congeners with superior antimicrobial properties. 11 Over 10.000 quinolones have been synthesized and about 20 have been launched including the most widely used ciprofloxacin and levofloxacin. 22 Quinolones consist of a bicyclic ring structure in which there is a substitution at position N-1, with various moieties. All the current agents have a carboxyl group at position 3, a keto group at position 4, a fluorine atom at position 6, and a piperazinyl group or a methylpiperazinyl group at the C-7 position. Differences in the moiety present at N-1 position or at C-7 position markedly influence both microbiological and pharmacokinetic properties. 9 The new drug application for clinafloxacin to the U.S. Food and Drug Administration (FDA) was withdrawn in 1999, the clinical use of several other quinolones, including gemifloxacin and trovafloxacin, was sharply limited, or have been withdrawn, eg, gatifloxacin, grepafloxacin, sparfloxacin and temafloxacin.23–26
In a study by Ball et al it has been reported that sparfloxacin and grepafloxacin, have prolonged QT in either animals or humans. 24
The abrupt withdrawal of grepafloxacin by its manufacturer, in association with possibly associated, sudden cardiac deaths, has again highlighted the phenomenon of quinolone-induced QT prolongation and the potential for associated ventricular tachy-arrhythmias originally described with sparfloxacin. 23 However, quinolone-related malignant ventricular tachy-arrhythmias have been described very rarely and only with sparfloxacin, levofloxacin and, most recently, grepafloxacin.23,24,27,28 In animal toxicological assessment, these agents have been proved to prolong QT interval.24,29,30 For trovafloxacin, withdrawn because of hepatotoxicity, but no animal data are available but the European CPMP commented on QT prolongation within normal physiological limits. 24
Sitafloxacin (also called Gracevit, DU-6859a, Figure 1) is a new-generation broad-spectrum oral fluoroquinolone that shows promise in the treatment of vast entities of bacterial infections, especially associated with multidrug-resistant bacteria.31–33 The structural nomenclature of sitafloxacin is unique: 7-[4(S)-amino-6-azaspiro[2,4]heptan-6-yl]-8-chloro-6-fluoro-1-[(1R,2S)-2-flurocyclopropyl]-4-oxo-1,4-dihdroquinoline-3-carboxylic acid which includes amino-pyrrolidine substituent at C-7 and a fluorocyclopropyl group at N-1. It contains a chlorine substituent at position 8 of the quinolone nucleus. 14 The antimicrobial compound was identified by Daiichi Sankyo Co., which brought ofloxacin and levofloxacin to the market.34,35 Sitafloxacin has been in commercial use in Japan since 2008 and exhibits higher antimicrobial activity to pathogenetic Gram-negative and Gram-positive bacteria than does either levofloxacin or ciprofloxacin.9,36
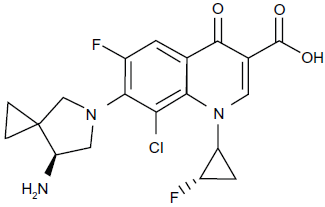
Chemical structure of sitafloxacin.
In Japan, it has been approved in 2008 for bacterial infections such as laryngopharyngitis, tonsillitis, acute bronchitis, pneumonia, secondary infections of chronic respiratory diseases (eg, AECOPD), cystitis, pyelonephritis, urethritis, cervicitis, otitis media, sinusitis and few oral infections: most seen in elderly people, although fluoroquinolone resistance among elderly patients appears to be increasing.5,31,37
Pharmacodynamic
Physico-chemical characteristics
Sitafloxacin has the properties of an ampholytic electrolyte due to the presence of a carboxyl and an amino group in its molecular structure; it shows a relatively low aqueous solubility (100 mg/mL), but higher solubility in organic solvents such as chloroform (3000 mg/mL). Thus, due to the solubility characteristics it was obviously important to develop and evaluate an oral formulation of sitafloxacin which could provide adequate oral bioavailability and plasma level profiles compatible with providing effective plasma antibacterial concentrations. 38
Mode of action
The bactericidal activity and mode of action of many quinolones have been described in earlier studies.39–49 Sitafloxacin inhibits DNA gyrase and topoisomerase IV. These enzymes are involved in bacteria DNA replication, transcription, DNA repair and recombination.11,50 Sitafloxacin has less activity against human topoisomerase II, an enzyme which is involved in cell growth. 51 According to Akasaka et al. 51 DNA gyrase, consisting of the subunits GyrA and GyrB, has ATP-dependent DNA supercoil-ing activity and is a primary target of quinolones in the Gram-negative species, such as Escherichia coli and Neisseria gonorrhoeae.11,14 In contrast, topoisomerase IV, consisting of the subunits ParC and ParE, has an essential role in partitioning replicated chromosomes and is more sensitive than DNA gyrase to some quinolones, such as levofloxacin and ciprofloxacin, in the Gram-positive species, such as Staphylococcus aureus and Streptococcus pneumoniae. 14
However, S. pneumoniae in vitro studies by Okumura et al, Onodera et al and Morrissey et al demonstrated that sitafloxacin has dual-activity in terms of inhibition of pneumococcal DNA gyrase and topoisomerase IV.50,52–55 Whereas other studies described as the primary target of sitafloxacin DNA gyrase in E. coli, in Mycobacterium tuberculosis, and topoisomerase in S. aureus.56–58
Antibacterial activity
Sitafloxacin has a high in vitro activity against Gram-positive and -negative bacteria, including atypical pathogens, mycobacteria species, and anaerobes.36,59–63 Based on a mean AUC (0-24 h) of 25 µg · h/mL for sitofloxacin, microbiological and clinical efficacy would be achieved by MIC ≤ 0.25 µg/mL for Gram-negative and ≤0.5 µg/mL for Gram-positive bacteria, revealing AUC/MIC breakpoints for sitafloxacin of 100 and 30, respectively. 38 Thus, combining the pharmacokinetic results of the present study with the MIC data from the large European surveillance study and applying the above mentioned pharmacodynamic breakpoints, sitafloxacin appears to be active against 100% of the three major respiratory tract pathogens (S. pneumoniae, H. influenzae, M. catharrhalis), 96% of staphylococci including methicillin-resistant strains, 90% of the enterobacterial strains, 70% of the enterococci including vancomycin-resistant strains and 63% of Pseudomonas strains.36,38 In vitro and in vivo activity of sitofloxacin is reviewed in the section Efficacies of sitafloxacin in different bacterial species.
Resistance
Resistance to fluroquinolones is mainly associated with mutations in the quinolone resistance-determining regions (QRDR) of the genes encoding DNA gyrase (gyrA and gyrB) and topoisomerase IV (parC and parE). 11 Mutations in genes encoding membrane-associated efflux pumps may also contribute to the development of fluoroquinolone resistance. 11 Alterations in GyrA seem to play an important role in resistance. However, sitafloxacin demonstrated in vitro activity against various mutant S. pneumoniae with gyrA or parC mutations and fluoroquinolone-resistant isolates with multiple mutations in gyrA, parC and/or parE.50,64,65
Pharmacokinetics
Absorption, serum protein binding and tissue distribution
Nakashima et al studied the pharmacokinetics of sitafloxacin among other quinolones in healthy Japanese volunteers who received oral doses of 50 or 100 mg, a mean maximum sitafloxacin serum concentration (Cmax) of 0.51 and 1 µg/mL was revealed after tmax of 1.2 hours. 66 The mean area under the serum concentration-time curve (AUC) from zero to infinity was 2.62 µg · h/mL (50 mg) and 5.55 µg · h/mL (100 mg), respectively. Thus, peak levels occurred about 1 hour after dosing. The peak concentration and AUC were roughly proportional to the drug dose. Food intake did not affect the pharmacokinetics of single-dose sitafloxacin 100 mg to a clinically significant extent. There was no evidence of drug accumulation after multiple dosing of sitafloxacin. Terminal half-life of sitafloxacin was 4.5-5 hours. The absolute oral bioavailability of sitafloxacin was 89%. After 1-8 hours of single 100 mg dose of sitafloxacin, the serum protein binding was 46%-55%. 66
Sasaki et al investigated the penetration of sita-floxacin into tissues and body fluids. Mean sitafloxacin concentrations were 0.57 µg/g in oral cavity and 0.32 µg/g in fluid extracted from the wound after 2.7-3.7 hours after administration of 50 mg sitafloxacin to odontoectomy patients with odontogenic infections. 67 Baba et al studied the efficacy and safety of sitafloxacin in Japanese otorhinolaryngological patients. In the mucous membrane of the middle ear, mean sitafloxacin concentrations were 0.82 µg/g versus 0.59 µg/g in serum, 0.56 µg/g in the mucous membrane of the maxillary sinus, whereas the highest concentration of sitafloxacin was determined in the mucous membrane of the ethmoid sinus with 0.96 µg/g, after 2-4 hours administration of single dose of 100 mg sitafloxacin. 68
Metabolism and excretion
For elimination of sitafloxacin metabolism plays only a small role. F-magnetic resonance spectroscopy suggested that sitafloxacin does not undergo retention in the liver when administered 500 mg/day for 5 days in healthy individuals.69,70
In vitro studies by the manufacturing company described modest inhibition of cytochrome P450 (CYP) enzymes CYP1A1 and CYP1A2, but no inhibition of the other enzymes (CYP3A4, CYP2C9 and CPY2D6).31,32 Studies in different animals indicate that sitafloxacin is rapidly absorbed and widely distributed into various tissues. Sitafloxacin-related material is eliminated primarily through both renal and biliary excretion in rats, and possibly in dogs, whereas renal excretion is the major route of elimination in monkeys. 69 In human, after 48 h post-dose, sitafloxacin (single dose 50 or 100 mg) sitafloxacin was excreted almost exclusively unchanged in the urine. 66
Sitafloxacin in elderly patients and those with severe illness
Comprehensive studies of sitafloxacin in the elderly patients have not been published yet in English journals with clinically significant changes in pharmacokinetic parameters such as AUC, clearance and Cmax. And such sufficient studies are needed Data on the pharmacokinetics of sitafloxacin in renal disease are currently limited; however with increasing severity of renal impairment, the sitafloxacin concentration might increase due to delayed urinary excretion. 71 Dosage adjustment is recommended in patients with creatinine clearance rates of 10-60 mL/min. In volunteers aged 67-80 years, as compared to younger volunteers (25-35 years) the mean AUC24, tmax and half-time of sitofloxacin increased, whereas Cmax decreased by one third. 72
Similar to alterations in renal function, changes in hepatic function can potentially affect the dosage adjustments of fluoroquinolones, particularly those agents with significant non-renal clearance mechanisms. Although robust data on sitafloxacin are missing, dosage adjustments in hepatic dysfunction are not likely to be required as this antibiotic due to its renal elimination. In a recent publication Nakajima et al evaluated microbiological and clinical effects of the systemic administration of sitafloxacin on periodontal pockets in elderly patients receiving supportive periodontal therapy. The presence of periodontal bacteria (Porphyromonas gingivalis, Treponema denticola and Tannerella forsythia) was significantly reduced at 1 month after treatment. 73
Adverse effects
Side effects of the quinolones have varied according to the specific compounds and include central nervous system stimulation, gastrointestinal disturbances, vasculitis, and photosensitization. The compound is characterized by a cis-oriented (1R,2S)-2-fluorocy clopropylamine moiety that is indispensable for its reduced side effects and superior pharmacokinetic profile.37,74–76 Matsumoto et al conducted a 2-yrs-survey of Japanese 3558 sitafloxacin-treated cases and analysed the efficacy and safety of this antimicrobial agent. The incidence of adverse reactions was 4.44% (148/3331 cases). Major adverse reactions were diarrhea (55 cases) and hepatic function disorders (39 cases), and the incidences were 1.65% and 1.17%, respectively. Serious adverse reactions were observed in 5 cases: gastrointestinal bleeding, abnormal hepatic function, decreased white blood cell count, drug eruption, hypoglycemia, pneumonia, and superinfection. The authors concluded that sitafloxacin demonstrated no serious problems in its safety profile. 37 According to Feldman et al, mild transient increases in alanine aminotransferase and alkaline phosphatase occurred in individuals treated with sitafloxacin, but there were no apparent trends in the other serum enzyme levels. 77
Fluoroquinolones are associated with dermal phototoxicty following exposure to sunlight. A randomized controlled trial (volunteer study) by Dawe et al was conducted in healthy Caucasians (n = 40) and Orientals (n = 17) and compared the phototoxic potential of sitafloxacin, with sparfloxacin, enoxacin, levofloxacin and placebo. In healthy Caucasians, sitafloxacin 100 mg twice a day produced mild ultraviolet (UV) A-dependent phototoxicity at 365 ± 30 nm, maximal at 24 h with normalization by 24 h post-drug cessation. In comparison, the sparfloxacin group experienced severe phototoxicity maximal at 24 h and extended in the visible region (430 ± 30 nm), maximal at 400 ± 30 nm with abnormal pigmentation at on-drug phototest sites lasting, although fading, for up to 1 year. Phototoxicity was not detected in the levofloxacin or placebo groups. In the Oriental study, no clinically relevant phototoxicity was seen with either sitafloxacin or placebo groups. 76 The authors conclude that 100 mg of sitafloxacin twice a day in Caucasians was associated with a mild degree of cutaneous phototoxicity. Enoxacin 200 mg three times a day and sparfloxacin 200 mg/day were much more photoactive. In the Oriental study group, which was limited by small numbers (n = 17), sitafloxacin 50 mg twice a day and 100 mg twice a day failed to demonstrate a clinically significant phototoxic effect. 76 Rash and seizures, probably associated with sitafloxacin, were observed in four patients by Shetty & Wilson. 78
Drug-drug interactions
Niki et al conducted in vitro and in vivo studies to investigate the drug interaction between a sita-floxacin (DU-6859a), and theophylline. Sitafloxacin administered at a dose of 50 mg resulted in no changes in serum theophylline concentrations, and slight increases in serum theophylline concentrations were observed at a dose of 100 mg. However, this was clinically not significant. Moreover, the administration of 100 mg of sitafloxacin resulted in decreases in all urinary theophylline metabolites. 79 Another pharmacon, ranitidine, did not affect the pharmacokinetics of sitafloxacin significantly. 80 Shiba investigated also the pharmacokinetic effects of metal-ions on sitafloxacin. Co-administration of aluminium hydroxide, magnesium oxide, calcium carbonate and ferrous sulphate reduced the sitafloxacin Cmax by 37%-82%, and similarly reduced AUC24 of sitafloxacin. 81
Nakajima et al studied the in vivo and in vitro antifungal activity of sitafloxacin in combination with antimycotic agents. Sitafloxacin combined with amphotericin B enhanced the in vitro antifungal activity of amphotericin B against Candida spp. and Cryptococcus neoformans. Positive interactions of sitofloxacin with amphotericin B against Aspergillus fumigatus were dependent on the medium used. Synergism was demonstrated in yeast nitrogen base supplemented with amino acids, ammonium sulfate, and 1% glucose, whereas antagonism between the drugs occurred in RPMI 1640 medium. In combination with fluconazole, sitofloxacin increased the activity of fluconazole against C. albicans both in synthetic amino acid medium-fungal and in supplemented yeast nitrogen base. Furthermore, in a mice model infected with C. albicans, the fungal load in the kidneys was significantly less in mice given the combination treatment of sitofloxacin plus either amphotericin B or fluconazole. The combination treatment resulted in prolonged survival of infected mice compared to the mice treated with either antimycotic agent alone. The prolonged survival in mice treated with sitafloxacin and amphotericin B was also observed in A. fumigatus infection, indicating that sitofloxacin enhanced the activity of the antimycotic agents in vivo as well as in vitro. 82
Efficacies of Sitafloxacin in Different Bacterial Species
Gram-positive bacteria
Methicillin-resistant Staphylococcus aureus (MRSA) poses a serious threat to worldwide health. Much focus over the past 50 years has been on hospital-associated MRSA (HA-MRSA).83–87 However, more than a decade ago, MRSA (community-associated, CA-MRSA) has spread into the community causing disease in otherwise healthy people with no discernible contact with healthcare settings and facilities. In contrast to the HA-MRSA, the CA-MRSA strains do not necessarily harbour the quinolone resistance genes.85–87,88 Although the relative role of widespread fluoroquinolone use and ineffective infection prevention and control practices in encouraging the spread of virulent Clostridium difficile-associated diarrhea in healthcare settings remain controversial, there are evidences that quinolone-driven colonization with MRSA enhances the risk of subsequent infection. 89 However, Chong et al observed that quinolone prophylaxis for neutropenia does not induce a significant increase in the growth of quinolone- and multidrug-resistant bacteria, but discontinuing quinolone prophylaxis may induce a dramatic increase in the growth of Gram-negative bacteria, including ESBL-producing pathogens. 90 Similarly, Laufarie et al showed that the reduction of quinolone use led to a significant decrease in quinolone-resistant MRSA rate. 91 Therapy of infections caused by resistant Gram-positive bacteria requires appropriate use of available antibiotics and stewardship to prolong their effectiveness. 92 Nosocomial MRSA strains displaying resistance to fluoroquinolones and acquiring additional resistance genes usually show compromised fitness and are not able to widely disseminate in the community where resistance to quinolones is not an asset and quinolone-sensitive CA-MRSA isolates could easily outgrow them. As one of the newer quinolones, sitafloxacin is active against staphylococci. 36 The MIC90 for oxacillin-resistant strains ranged from 0.25 to 1 µg/mL and were considerably higher than those for oxacillin-susceptible strains (MIC90, 0.03 to 0.12 µg/mL), as shown for other quinolones (Table 1).93–96 Sitafloxacin was the most active agent against oxacillin-resistant strains; its activity was twice or more higher than that of clinafloxacin and at least eight times more active than the other quinolones. Of the 457 MRSA isolates tested, 95.4% were resistant to ciprofloxacin, whereas only 0.4% of the MRSA isolates were resistant to clinafloxacin and 0.2% resistant to sitafloxacin (MIC ≥ 4 µg/mL). 36 Earlier study by Giamarellou-Bourboulis et al demonstrated extended post-antibiotic and a rapid killing effect of sitafloxacin on MRSA resistant to ciprofloxacin and to rifampin, thus supporting their once daily administration in the therapy of infections by multiple drug-resistant MRSA. 97
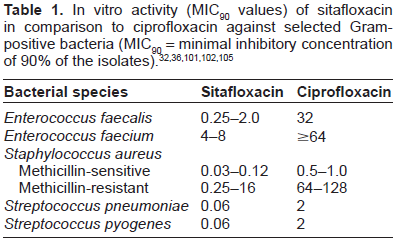
As reported for other quinolones, the activity of sita-floxacin against enterococci was generally lower than that against other Gram-positive cocci (staphylococci, streptococci.36,94,98 For E faecalis, the MIC50 and MIC90 were 0.12 and 2 µg/mL, respectively. The in vitro activity of sitafloxacin was at least four times higher than that of the other fluoroquinolones. The MICs of sitafloxacin for E. faecium were higher (MIC50, 0.5 µg/mL; MIC 90 , 4 µg/mL). For vancomycin-susceptible and -resistant enterococci, the MICs of all quinolones tested were comparable. Similarly, other studies confirmed the in vitro activities of sitafloxacin against MRSA and multiresistant enterococci.78,99–101 Shetty & Wilson evaluated in a phase II non-comparative trial the activity of sitafloxacin (400 mg iv over 1 h once a day for 7-14 days) in patients with serious systemic infections with MRSA or vancomycin-resistant enterococcus (VRE). There were 10 infections with MRSA alone, eight with VRE alone and one with MRSA plus VRE. 101 Four out of 11 MRSA-patients, unresponsive to glycopeptides, were cured, six failed treatment and one was indeterminate. Five out of nine VRE-patients (one patient had both pathogens, MRSA and VRE), were cured and four failed. 111 Similar efficacy results for sitafloxacin in MRSA and enterococci were revealed in in vivo and in vitro studies by Kanda et al and in vitro studies by Yamaguchi et al as compared with other fluoroquinolones.32,102 Yamaguchi et al reported the results of in vitro susceptibilities of 12,919 clinical isolates from Japanese centers to selected antibiotics including sitafloxacin. 32
According to Milatovic et al sitafloxacin was also active against Streptococcus pneumoniae and the other Streptococcus spp. tested, with MIC90 ranging from 0.03 to 0.12 µg/mL. 32 Milatovic et al observed no difference between penicillin-susceptible and -resistant isolates, as was also shown by Visalli et al. 103 In S. pneumoniae, sitafloxacin was the most active agent, followed by clinafloxacin, trovafloxacin, moxifloxacin, gatifloxacin, levofloxacin, and ciprofloxacin. Browne et al studied the antipneumococcal activity of DK-507k, sitafloxacin and other quinolones in 26 quinolone-resistant pneumococci with known resistance mechanisms. 104 DK-507k and sitafloxacin were the most active quinolones (MICs, 0.125-1.0 µg/mL), followed by moxifloxacin, gatifloxacin, levofloxacin, and ciprofloxacin. Mutations in quinolone resistance-determining regions (QRDR) of quinolone-resistant strains were in the usual regions of the parC and gyrA genes. Exposure to DK-507k and sitafloxacin led to mutations, mostly in gyrA.
In the study by Amano et al the in vitro activity of sitafloxacin and various oral antimicrobial agents against 1,620 bacterial isolates from different healthcare facilities in Japan was evaluated. 101 The MIC90 of sitafloxacin was 0.06 µg/mL for methicillin-susceptible S. aureus and was equal to that of garenoxacin, but lower than that of moxifloxacin and levofloxacin. Inhibitory activity of sitafloxacin in growth of S. pneumoniae isolates was at 0.06 µg/mL or less. The MIC90 of sitafloxacin ranged from 0.03 to 0.06 µg/mL and was lower than MIC90 of garenoxacin, moxifloxacin, and of levofloxacin with increasing factor. Similarly, the MIC90 of sitafloxacin was 0.06 µg/mL for Streptococcus pyogenes, which was lower than that of other tested quinolones (Table 1). The MIC90 of sitafloxacin for E. faecalis was higher (0.25 µg/mL), but still lower than that of the comparator quinolones. 101
Gram-negative bacteria
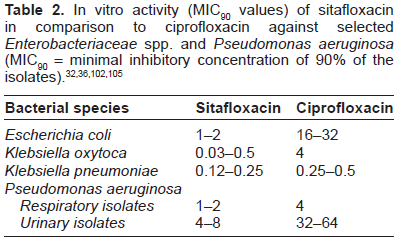
In a comprehensive study by Milatovic et al the in vitro activity of sitafloxacin in comparison to other quinolones against Gram-negative and -positive bacteria, and anaerobes was determined. 32 The MIC of sitofloxacin for Enterobacter aerogenes and Escherichia coli was 1 µg/mL for each. These species also exhibited the highest rates of ciprofloxacin resistance. The MIC90 of sitafloxacin for Citrobacter koseri, Klebsiella oxytoca, Pantoea agglomerans, Proteus vulgaris, Salmonella spp., Serratia liquefaciens, Shigella spp., and Yersinia enterocolitica ranged from 0.015 to 0.12 µg/mL and were similar to those of ciprofloxacin. Moreover, sitafloxacin was four times more active than ciprofloxacin against Citrobacter freundii, Klebsiella pneumoniae, Morganella morganii, and Serratia marcescens and at least eight times more active against E. aerogenes, E. cloacae, E. coli, and Proteus mirabilis. 32 Amano et al also evaluated sitofloxacin efficacy against Gram-negative bacteria. 101 The MIC90 of sitafloxacin for E. coli was 2 µg/mL, and the MIC90 of other 10 species of Enterobacteriaceae which were the lowest values of the quinolones tested ranged from 0.03 to 1 µg/mL (Table 2).
A randomized open-label multicenter phase II study of sitafloxacin (400 mg) versus imipenem/cilastatin (500 mg three times per day) has been processed in South Africa patients with pneumonia associated with Gram-positive and -negative bacteria. 77 In both groups clinically evaluable patients were cured in more than 90%. In bacteriologically evalu-able patients, 90%-95% of the sitafloxacin patients had acceptable bacteriological response whereas the response of the imipenem/cilastatin group was 100%. For all patients in both treatment groups, no pathogens were isolated at the first follow-up assessment at 24-72 h after treatment. All of the pathogens tested in the sitafloxacin treatment group were sensitive to that antibiotic and all pathogens tested in the imipenem treatment group were sensitive to that antibiotic. Of the tested pathogens in the imipenem treatment group, all were sensitive to sitafloxacin. Feldman et al concluded according to these data that sitafloxacin can be considered to show safety, tolerability and perhaps comparable efficacy as imipenem/cilastatin. 77
Okuda et al evaluated the in vitro activity of sita-floxacin using clinical isolates of Vibrio cholerae O1 in comparison to other fluoroquinolones: ciprofloxacin, ofloxacin, sparfloxacin and levofloxacin. Against fluoroquinolone-susceptible V. cholerae O1, the MIC90 of sitafloxacin was 2- to 4-fold lower than the other fluoroquinolones. This suggests sitafloxacin might be used in the treatment of infections caused by V. cholerae O1 strains including the fluoroquinolone-resistant strains. 106
According to Bale et al, Jones et al and Milatovic et al, sitafloxacin demonstrated high efficacy against Haemophilus influenzae, Moraxella catarrhalis, Neisseria meningitidis, and Neisseria gonorrhoeae, the MIC90 being ≤0.008 µg/mL for all species, including β-lactamase-positive strains.36,107,108 The sitafloxacin MICs for five gonococcal strains resistant to ciprofloxacin ranged from 0.03 to 0.12 µg/mL. Sitafloxacin inhibited the growth of Haemophilus influenzae at 0.004 µg/mL or less, and of Moraxella catarrhalis at 0.008 µg/mL, both MIC90 values were lower than those of the other quinolones. 101
Another Gram-negative pathogen which might be also related to chronic infections in the elderly people is Helicobacter pylori. Over the last decades, eradication rates in first-line H. pylori therapy have been declining, mainly due to increasing resistance against the recommended antibiotics clarithromycin and metronidazole. Based on a large number of studies, quinolones have been introduced in second-line and rescue treatment and are recommended for these indications in current guidelines.60,109 Various studies have investigated alternative strategies for first-line treatment including quinolone-based regimens. In the context of increasing resistance rates of H. pylori against quinolones some risks and benefits have to be considered when using quinolones as a first-line strategy. Besides numerous studies investigating levofloxacin and moxifloxa-cin there are some promising results for sitafloxacin. Sitafloxacin might overcome primary resistance of H. pylori against conventional quinolones. It has been reported to have a high antimicrobial activity against H. pylori, and has been suggested for use in therapy for patients who have failed H. pylori eradication using traditional therapies.60,110,111
Nonfermenters
Resistance to quinolones may occur generally in Gram-negative nonfermenters.36,98,112 Sitafloxacin was at least eight times more active than ciprofloxacin against Acinetobacter spp., Burkholderia cepacia, and Stenotrophomonas maltophilia. Almost complete cross-resistance between ciprofloxacin and sitafloxacin was observed in Pseudomonas aeruginosa, with only 5% of the 165 ciprofloxacin-resistant isolates being susceptible to sitafloxacin at ≤1 µg/mL. The study results of Kitamura et al led to the conclusion that DU-6859a is much more active against quinolone-resistant clinical isolates of P. aeruginosa than other quinolones, probably because of its strong inhibitory effects against mutant quinolone-resistant DNA gyrases, and that the C-8 chlorine is necessary for these potent effects. 113 A study by Otani et al demonstrates the results of comparison between sitafloxacin DK507k, and ciprofloxacin. 114 Sitafloxacin showed less phototoxicity than DK507k and was more active against Pseudomonas spp., and the MIC90 value was lower than that of ciprofloxacin and DK507k. 114 Amano et al reported that the MIC of sitafloxacin for urinary isolates of P. aeruginosa was 8 µg/mL which was 16 times lower than the MICs of the other quinolones, whereas the MIC of sitafloxacin for P. aeruginosa respiratory isolates was lower (2 µg/mL) and was 16-32 times lower than the MICs of the other quinolones (Table 2). 101
Mycobacteria
Tuberculosis (TB) is an important public health problem worldwide due to AIDS epidemic, the advent of multidrug resistant strains (MDR) and the lack of new drugs in the market. Thus, there is an urgent need for new drugs or combination of currently available drugs to combat this disease. Due to the importance of the quinolones in the fight against tuberculosis, sitafloxacin has also been under preclinical and clinical study. The 50% inhibitory concentration (IC50) of sitafloxacin was much lower than that of the comparator quinolones against supercoiling activity of DNA gyrase. 57
Sitafloxacin and gatifloxacin exhibited much more potent anti-M tuberculosis activity than levofloxacin did. Notably, sitafloxacin and gatifloxacin were eight times more active against MDR-M. tuberculosis strains than levofloxacin in terms of MIC50, suggesting that these new quinolones might be useful in clinical control of MDR-M tuberculosis. 62
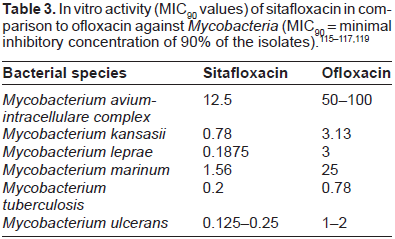
The antimicrobial effect of sitafloxacin against other Mycobacterium species, such as Mycobacterium leprae, was studied by Dhople & Namba, either alone or in combination with either rifampicin or rifabutin. 116 The results were compared with those obtained with ofloxacin. When used singly, sitafloxacin and ofloxacin inhibited the growth of M. leprae completely at MIC values 25 and 100 mg per kg body weight per day, respectively, and the effects were bactericidal. Both sitafloxacin and ofloxacin exhibited excellent synergistic effects when combined with either rifabutin, but not with rifampicin. When compared to other quinolones, sitafloxacin was found to be more potent than the other three commonly used fluoroquinolones (ciprofloxacin, levofloxacin and ofloxacin), with MIC = 0.1875 µg/mL against M. leprae (Table 3). 116 The MIC values of sitafloxacin for M. ulcerans were in the range of 0.125-0.5 µg/mL, much lower than that of other quinolones. 117 Sitafloxacin showed favourable antimycobacterial activity in vitro and in vivo, as compared with moxifloxacin against Mycobacterium avium infection in mice. Although moxifloxacin exhibited the strongest activity against intramacrophage M. avium, when each test quinolone was administered alone to infected mice, sitafloxacin and gatifloxacin exhibited greater therapeutic efficacy than moxifloxacin based on intrapulmonary bacterial elimination. However, moxifloxacin exerted greater activity in killing bacteria in the spleen. Sitafloxacin and moxifloxacin exhibited combined effects on intrapulmonary bacterial elimination when administered to mice in combination with clarithro-mycin plus ethambutol. Sitafloxacin exerted the most marked combined effects in bacterial killing in the spleen. Levofloxacin displayed the lowest in vitro and in vivo activities amongst the tested quinolones. In conclusion, these findings indicate that sitafloxacin and moxifloxacin exhibit favourable activities against M. avium in vitro and in vivo. 118
Intracellular pathogens
Miyashita et al investigated the in vitro and vivo activity of sitafloxacin against Chlamydia spp. in comparison to other quinolones, including ciprofloxacin, ofloxacin, sparfloxacin, and tosufloxacin. The chlamydial species tested were C. psittaci, C. trachomatis and Chlamydophila pneumoniae. 59 These bacterial species are well-known respiratory pathogens that cause upper and lower respiratory tract infections and pneumonia. The zoonotic pathogen C. psittaci is responsible for psittacosis, C. trachomatis for pneumonia in neonates and Chlamydophila pneumoniae is among the most common pathogens for community-associated pneumonia. Some of the previously developed quinolones have shown excellent antichlaymdial activity in clinical studies, in vitro and in vivo (Table 4).120–125 Sparfloxacin was the most active compound tested, followed by ofloxacin and temafloxacin. Ciprofloxacin and fleroxacin were the least active. 120
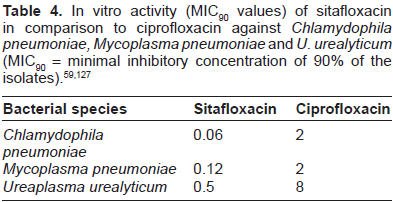
In the study by Miyashita et al, sitafloxacin showed good in vitro antichlamdial activity equal or superior to that of sparfloxacin, tosufloxacin, ofloxacin, and ciprofloxacin. The MICs of sitafloxacin for the different species ranged from 0.031 to 0.125 µg/mL. The authors discussed the different activities of the quinolones and presumed that the differences may be due to the different pharmacokinetics of the compounds. The AUC was the lowest for sitafloxacin (0.34). From these results the authors concluded that the in vivo potency seen in their study was not due to a better pharmacokinetic profile. 59
Another pathogen associated with acute non-gonococcal urethritis or urogenital infections, besides C. trachomatis, is Mycoplasma genitalium. There is no precise clinical recommendation for M. genitalium induced cervicitis, although M. genitalium is one of the pathogens responsible for uterine cervicitis. 126 Terada et al retrospectively investigated the antimicrobial efficacies of several antibiotics against cervicitis caused by M. genitalium. A total of 257 women with M. genitalium associated cervicitis, except for those with chlamydial and gonococcal infections, were treated with one of the following antibacterial agents: azithromycin, clarithromycin, moxifloxacin, levofloxacin, and sitafloxacin. PCR-based assay was performed to evaluate the microbiological efficacy of eradication. M. genitalium was eradicated from the cervix in 90.5% of the patients treated with azithromycin (2 g single dose) and those treated with moxifloxacin (400 mg/day for 7 days). The pathogen was eradicated in all patients (100%) treated with moxifloxacin (400 mg/day for 14 days). However, M. genitalium was eradicated in 12 of the 13 (92.3%) patients treated with sitafloxacin (200 mg/day for 14 days). The authors concluded that azithromycin, moxifloxacin, and sitafloxacin would each be an effective treatment for M. genitalium infection. 126
Anaerobes
Ciprofloxacin and ofloxacin as the first generation of fluoroquinolones are inactive against most anaerobic bacteria. However, some broad-spectrum quinolones, which have recently become clinically available, have significant antianaerobic activity. Fluoroquinolones with increased in vitro activity against anaerobes include levofloxacin, clinafloxacin, sparfloxacin, trovafloxacin, grepafloxacin, moxifloxacin and sitafloxacin.14,74 Quinolones with intermediate antianaerobic activity include sparfloxacin and grepafloxacin. Trovafloxacin, gatifloxacin and moxifloxacin yield low MICs against most groups of anaerobes. Quinolones with the greatest in vitro activity against anaerobes include clinafloxacin and sitafloxacin. 14 Nord et al investigated the antimicrobial activity of sitafloxacin versus other quinolones in different anaerobic bacteria, including peptostreptococci, Clostridium perfringens, Clostridium difficile, Bacteroides fragilis, Porphyromonas, Prevotella, and Fusobacterium (Table 5). 61 The in vitro activity of sitafloxacin was superior to those of comparator quinolones. Kato et al compared the in vitro activity of sitafloxacin (DU-6859a) with those of other fluo-roquinolones against clinical isolates of anaerobic bacteria and Gardnerella vaginalis. 129 Sitafloxacin was the most active agent; it inhibited 90% of isolates of almost all species tested, including Bacteroides fragilis <0.39 µg/mL. Although the other quinolones tested were active against Gram-positive anaerobes, inhibiting their growth at <1.56 µg/mL, these agents were less active against the B. fragilis group and Prevotella bivia (90% of which were inhibited at >6.25 µg/mL). Mobiluncus species and G. vaginalis were inhibited by sitafloxacin at 0.1 µ/mL. According to Kato et al, sitafloxacin is a promising oral agent for the treatment of bacterial infections due to anaerobic bacteria; however, further studies are obligatory concerning the effects of this compound in bacterial vaginosis. 129
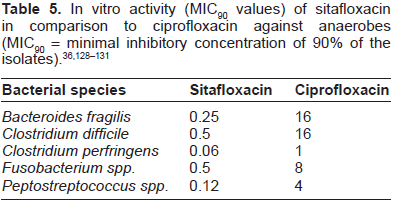
Spangler et al studied the activities of sitafloxa-cin (DU-6859a) and other quinolones, including ciprofloxacin, levofloxacin, sparfloxacin, and other antibiotics, eg, piperacillin, piperacillin-tazobactam, imipenem, clindamycin, and metronidazole against 11 anaerobes. 131 Sitafloxacin was the most active drug tested (MIC: 0.06-0.5 µg/mL), followed by imipenem (MIC: 0.002-4.0 µg/mL). All compounds were bactericidal at the MIC after 48 h; after 24 h, 90% killing was shown for all strains when the compounds were used at four times the MIC. DU-6859a at ≤0.5 µg/mL was bactericidal after 48 h. According to Goldstein et al, gemifloxacin was tested against 359 clinical anaerobic isolates and its activity was compared with other antimicrobials, including trova-floxacin, levofloxacin, grepafloxacin, sparfloxacin, sitafloxacin, penicillin G, metronidazole etc. 132 While all the comparative antibiotics were less active than gemifloxacin, sitafloxacin was equivalent to gemifloxacin against peptostreptococci, C. perfringens, and C. ramosum, and sitafloxacin was 2 to 3 dilutions more active against fusobacteria. The MICs of sitafloxacin ranged from 0.015 to 0.12 µg/mL for the species of anaerobic bacteria and were the lowest values of all the antimicrobial agents tested. 101 Clostridium difficile infection has become one of the most significant threats to hospitalised patients and represents an increasingly important issue in terms of morbidity and mortality within health care facilities. Ackermann et al analyzed the antimicrobial susceptibility of 80 clinical isolates of C. difficile. Nineteen strains that were found to be highly resistant to moxifloxacin (MIC ≥ 16 µg/mL) were then selected for extended antimicrobial susceptibility testing. All C. difficile isolates were inhibited by 2 µg/mL of sitafloxacin; among the other quinolones, only clinafloxacin showed similar activity. 133
Summary
Sitafloxacin is a new-generation, broad-spectrum oral fluoroquinolone that is very active against Gram-positive, -negative and anaerobic clinical isolates, including strains resistant to other fluoroquinolones, and has been approved in Japan for the treatment of respiratory and urinary tract infections in 2008. The mechanism of action of sitafloxacin, comparable to the majority of fluoroquinolones, involves the prevention of bacterial DNA synthesis, by inhibition of DNA gyrase, an enzyme required for the replication of DNA and of topoisomerase IV. Sitafloxacin, together with moxifloxacin, belongs to the same generation of fluoroquinolones and are called respiratory quinolones. They possess improved activity against Gram-positive bacteria and anaerobes located within a biofilm or intracellularly, and they demonstrated good tissue penetration. Among the Gram-positive and -negative, including anaerobic pathogens the in vitro antibacterial activity has been demonstrated against clinical isolates of S. aureus, including MRSA, and S. epidermidis, all strains of S. pneumoniae, S. pyogenes; P. aeruginosa, H. influenzae, M. catarrhalis, N. gonorrhoeae, the anaerobic bacteria, including Bacteroides fragilis, Clostridium spp., Fusobacteriuam spp. Peptostreptococcus spp., and the Enterobacteriaceae. Sitafloxacin has also been shown to be effective against pneumonia caused by Legionella pneumophila, S. pneumoniae, Mycoplasma pneumoniae and P. aeruginosa in animal models. Its activity against Gram-positive cocci was comparable or superior to those of garenoxacin, moxifloxacin and levofloxacin. Due to its favorable pharmacokinetic profile, excellent in vitro potency, and in particular its enhanced activity against Gram-positive organisms and anaerobes, sitafloxacin clearly has potential as a useful agent for the treatment of a variety of infections. Clinical trials and susceptibility studies indicate that sitafloxacin might be efficient in treating infections of the respiratory and genitourinary tract, as well as in treating intra-abdominal and skin and soft tissue infections caused by Gram-positive cocci. The third-line treatment regimen including sitafloxacin is being considered for H. pylori after failure of clarithromycin- and metronidazole-based therapies, since it is in vitro active against H. pylori. Regarding the safety of sitafloxacin, few studies reported adverse reactions associated with sitafloxa-cin (eg, diarrhea, hepatic disorder, gastrointestinal bleeding etc.) whereas other studies concluded that sitafloxacin is an antibacterial agent with no serious problems in its safety profile and efficacy rates of over 90% against all infections. Daiichi has stated that it is believed to be inferior to its levofloxacin product, for which overseas data show the occurrence of photosensitivity adverse effects, especially in Caucasian population, although phototoxicity is essentially irrelevant if sitafloxacin is used in hospitals and especially in intensive care units. Therefore, Daiichi is planning for an IV formulation to be indicated only in patients with severe infections or drug-resistant diseases in markets outside Japan. No adverse effects have been reported in Japanese studies, believed to be most likely due to racial and dosage differences between patients overseas and in Japan. For Japanese patients, sitafloxacin provides the broad-spectrum coverage promised by clinafloxacin and trovafloxacin and comparable to carbapenems. Although sitafloxacin has no antifungal properties on its own, studies indicated that it enhanced the activity of currently available antifungal agents and might be useful in the treatment of Candida albicans infections.
In brief, sitafloxacin, as a new broad-spectrum quinolone, is a promising antimicrobial agent against several bacterial species including mycobacteria. However, more comprehensive in vitro studies, clinical trials on its safety and efficacy, especially in the elderly population in different regions other than in Japan should be conducted to rather reveal the phar-macodynamic and -kinetic parameters, the adverse effects and its reaction in co-morbidities within these patient groups.
Author Contributions
Conceived and designed the experiments: BG. Analysed the data: BG. Wrote the first draft of the manuscript: BG. Contributed to the writing of the manuscript: BG. Agree with manuscript results and conclusions: BG. Jointly developed the structure and arguments for the paper: BG. Made critical revisions and approved final version: BG. The author reviewed and approved of the final manuscript.
Funding
Author(s) disclose no funding sources.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest. Provenance: the authors were invited to submit this paper.