
Abstract
Partial seizures are frequently resistant to pharmacologic treatment. There are a plethora of medications currently approved for use in partial epilepsy. However, despite the large number of medications available, seizure control often remains elusive. A new medication with a unique mechanism of action has recently been approved for the adjunctive treatment of partial seizures in adults. Ezogabine (retigabine) exerts its actions at the level of voltage-gated potassium channels. In clinical trials it has demonstrated efficacy similar to that of other agents approved for resistant partial epilepsy. Adverse events tend to be central nervous system related. However, significant urinary retention has been noted in some persons exposed to the drug. A Risk Evaluation and Mitigation Strategy is being implemented to minimize urinary adverse events. Ezogabine represents an important addition to the partial epilepsy medication arsenal.
Introduction
Of all seizure types, partial seizures tend to be among the most difficult to control with pharmacotherapy. Partial seizures that remain localized account for approximately 15% of all seizures, while another 60% begin as partial seizures, and then spread to become secondarily generalized. 1 Simple partial seizures do not result in alterations of consciousness, but may be associated with motor symptoms, hallucinations and alterations of autonomic nervous system function. Complex partial seizures do result in impairment of consciousness, may be preceded by an aura, and often produce automatisms specific to an individual. 2 Consequences associated with uncontrolled partial seizures, particularly complex-partial seizures, may include a negative impact on overall quality of life, physical injury occurring during episodes of altered consciousness, permanent cognitive impairment and sudden unexpected death in epilepsy (SUDEP). 3
Medications Approved for Partial Seizures
There are a number of medications approved by the FDA as either monotherapeutic or adjunct options for the treatment of partial seizures. Older antiepileptic drugs, including phenytoin, valproic acid, carbamazepine and phenobarbital are designated monotherapeutic options. Newer antiepileptics that share this designation include topiramate, lamotrigine, oxcarbazepine and felbamate. Other medications approved by the FDA as adjunctive therapy for partial seizures that are not controlled using approved monotherapeutic options include gabapentin, pregabalin, tiagabine, levetiracetam, lacosamide, zonisamide and vigabatrin. When considering individual response rates (defined as at least 50% reduction in seizure frequency in clinical trials), most agents approved for monotherapy demonstrate similar rates of efficacy ranging from approximately 20%–50%. 4
Many of these drugs share similar mechanisms of action.1,5 Voltage-gated sodium channels are a frequent target, particularly of those medications considered to have a broad range of activity across the antiepileptic spectrum. Inactivation of sodium channels results in the inability to conduct sodium ions. Some antiepileptics inhibit sodium channels by binding to the channel in the open, depolarized state. The refractory period is prolonged, which ultimately blocks the sustained firing of neurons. Phenytoin, carbamazepine, oxcarbazepine, and lamotrigine are examples of drugs that work with relative exclusivity via fast inactivation. In contrast, lacosamide facilitates slow inactivation of sodium channels, and is the only drug to do so.
Voltage-gated calcium channels are also a frequent target of seizure-suppressing therapy. Gabapentin and pregabalin are believed to exert anti-epileptic activity via binding to the α2-δ protein found in auxiliary subunits of the voltage-gated calcium channel pore. This action is thought to decrease calcium influx resulting from depolarization, a phenomenon that normally results in neurotransmitter release. Drug binding at this site may regulate the release of those neurotransmitters, particularly excitatory glutamate.
GABAergic mechanisms are also common amongst antiepileptic drugs used for partial seizures. This is logical given the designation of GABA as the primary inhibitory neurotransmitter in the central nervous system (CNS). Allosteric changes at the GABAA binding site occur with felbamate (which also displays NMDA receptor antagonism), topiramate (also an antagonist at the AMPA/kainate subtype of the glutamate receptor) and phenobarbital. Tiagabine and vigabatrin exert unique GABA activity by inhibiting reuptake and decreasing metabolic breakdown of GABA respectively.
Several medications share multiple mechanisms of action, or possess activity that has yet to be fully defined. The broad-spectrum agent zonisamide is believed to possess activity at both sodium and calcium channels, and also appears to have some effect on GABA. Similarly, valproic acid is believed to have activity via these three sites of action. Levetiracetam on the other hand possesses a mechanism that is unique among the class in that it appears to exert its action via binding to a specific synaptic vesicle protein (SV2A) that is thought to play a role in neurotransmitter release.1,5
Given the resistant nature of partial seizures in many patients, and the overlap of mechanisms of action of many of the agents used to treat partial epilepsy, the development of agents with unique targets of action is a primary goal within the realm of antiepileptic therapy.
Ezogabine
Ezogabine (known internationally by the non-proprietary name retigabine) represents an entirely new mechanism of seizure inhibition. The drug was approved for adjunctive treatment of partial seizures in patients at least 18 years of age by the FDA on June 10, 2011, but is not yet available for use. (European approval was also granted earlier in the year). The official US launch has been delayed while the Drug Enforcement Administration assigns a schedule to the drug. It is anticipated that ezogabine will be marketed as a controlled substance since early data showed healthy subjects described feelings of euphoria (8.5%) and “feeling drunk” (0.9%) after ingesting single doses of the drug. 6 In phase 2 and 3 clinical trials, euphoric mood was reported by less than 1% of patients, however, recreational sedative-hypnotic users reported euphoria statistically similar to that experienced when under the influence of the benzodiazepine alprazolam. 6
Mechanism of action
Ezogabine is a derivative of the analgesic flupirtine, a centrally acting non-opiate pain medication that has been available in Europe since the mid-1980s. 7 It is not believed to exert any anti-seizure activity via sodium or calcium channels, nor does it appear to have significant activity related to glutamate or NDMA receptors. The potentiation of GABA has been observed in much higher concentrations compared to those employed in clinical trials to suppress seizure activity, but it is not believed that this mechanism plays a role in the antiepileptic activity of the drug at regular doses. 8
Ezogabine has sometimes been referred to as a “potassium (K+) channel opener”, but this is a misnomer. 9 Ezogabine binds to the hydrophobic pocket of the activation gate within the pore of voltage gated K+ channels (Kv7.2 and Kv7.3). Mutations of the genes corresponding to these channels, KCNQ2 and KCNQ3, have been identified as associated with benign familial convulsions and other idiopathic types of epilepsy.9–13 This family of channels is associated with the outward K+ current that causes repolarization of the membrane post action potential, also known as the M-current. 9 Patients are susceptible to epileptic activity when this current is inhibited. Ezogabine is believed to exert its primary anti-seizure effect by enhancing the M-current. The binding of ezogabine to the K+ channels shifts the action potential toward a hyperpolarized state producing an increase in the K+ current close to that of the normal resting potential.14,15
The drug's activity has been likened to bending the gate's “hinge” resulting in the gate remaining slightly open, thereby decreasing the angle through which the gate swings, and therefore the amount of energy needed to open it.7,9 In other words, ezogabine binding ultimately results in the need for a smaller depolarizing shift to open the channel, an increase in the speed at which the channel opens, and a prolongation of the open state of the channel (up to 4-fold). 16 Taken together, these effects result in an inhibition of repetitive firing.
Pharmacokinetics and drug interactions
The absolute bioavailability of an oral dose of ezogabine is approximately 60%. 17 Absorption is relatively rapid with peak serum concentrations reached within 1–2 hours. The drug displays linear pharmacokinetics up to doses as high as 1200 mg daily.18,19 Eight-hour dosing intervals are suggested as reflected by the single dose half-life (7.4–9.2 hours) and the half-life observed at steady state (approximately 7 hours). Food delays absorption, but does not affect the extent of absorption. 18
Up to 84% of ezogabine and its primary metabolites are excreted in the urine. 17 A decreased rate of renal ezogabine clearance has been noted in the elderly, and adjustments in dose are recommended. 17 Older subjects have demonstrated AUCs of up to 1.5 times that of younger subjects, and the drug's half-life may be prolonged by as much as 30%. 17 Ezogabine's primary metabolic pathways include acetylation and glucuronidation. 20 The drug does not appear to undergo metabolism via the cytochrome P-450 system, thus decreasing its propensity to interact with other medications. Ezogabine does have a partially active N-acetyl metabolite, and it too displays linear pharmacokinetics. 18 The contribution of this metabolite to the anticonvulsant activity of the drug is believed to be minimal. 21 Dose adjustments are suggested in severe hepatic disease.
The majority of other antiepileptic agents do not appear to result in significant drug interactions with ezogabine during concomitant use. 22 However, an interaction with lamotrigine has been identified in a group of 29 healthy male subjects exposed to both medications. 23 The AUC and the half-life of ezogabine were increased by 7.5% (P = 0.045) and 15% (P = 0.006) respectively. The AUC and half-life of lamotrigine were decreased (by 18% and 15%, P = 0.001 for both). Renal clearance of lamotrigine was also significantly increased. The decrease in lamotrigine parameters was unexpected given that ezogabine is not considered to be either a potent enzyme inducer or inhibitor. The clinical relevance of this interaction remains to be defined, but it is not believed that it will be of great clinical significance. Other medications, including propofol, valproic acid, lamotrigine, phenobarbital, imipramine and a combination oral contraceptive, have not demonstrated clinically significant drug interactions when given with ezogabine.15,24,25
Clinical efficacy trials
Data from three major trials of ezogabine were submitted to the FDA as evidence of the drug's efficacy in refractory partial seizures and ultimately led to the medication's approval (Table 1).
Summary of studies evaluating ezogabine efficacy in adult patient with partial epilepsy.
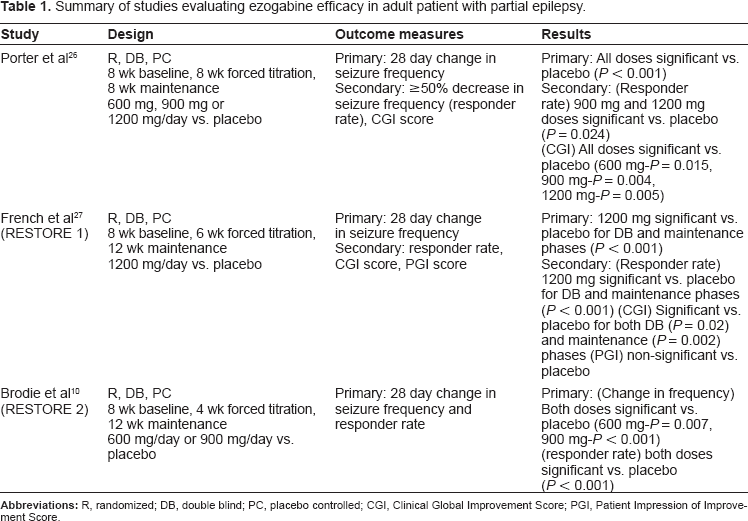
The first trial, conducted by Porter et al, was a randomized, placebo-controlled, dose-ranging trial of 396 patients (included in the intention to treat efficacy analysis) with partial seizures. 26 After an eight week baseline period, during which individuals 16–70 years of age were determined to be eligible for the trial based on each having had a minimum of four seizures per month, subjects were randomized to receive 300 mg, 600 mg or 900 mg of ezogabine daily in three divided doses, or placebo. Subjects either had a diagnosis of simple-partial (SP) seizures with a motor component (14.9%), complex-partial (CP) seizures with or without secondary generalization (86.9%), or both. Concomitant, constant-dose antiepileptic medications were allowed, and 70% of study participants were receiving the maximum two drugs in addition to the study medication. Concurrent therapies were represented by carbamazepine (54.2%), lamotrigine (29.2%), valproic acid (24.9%), topiramate (21.9%), gabapentin (17.3%), phenytoin (14.9%), oxcarbazepine, benzodiazepines and barbiturates. Vagal nerve stimulation therapy was allowed as long as the initial parameters did not change at any time during the study. Forced titration of each dose occurred over 8 weeks beginning with a dose of 100 mg three times daily and increasing by 50 mg/dose at weekly intervals. If intolerance occurred during the titration phase, two separate dose reductions of 100 mg/day were allowed. After titration, no dose changes were allowed during the ensuing maintenance phase (also 8 weeks long). The primary outcome was the change in overall seizure frequency per 28 days. Secondary outcome measures included the responder rate (≥50% decrease in seizure frequency) and a clinical global improvement (CGI) score. Seizure rates were reported via subject diaries. The CGI score was measured by physicians at the end of the 16-week trial utilizing a seven point scale ranging from “very much improved” (a score of 1) to “very much worse” (a score of 7). All doses of active study medication achieved clinically significant decreases in median seizure frequency compared to placebo (23% (600 mg), 62% (900 mg), 50% (1200 mg), P < 0.001 for all). However, in a pair wise comparison, the 600 mg dose was not found to be statistically superior to placebo. The mean change in seizure frequency in the 600 mg group actually increased by 8.57%, however this was attributed to significant outliers. Seizure reductions of 50% or greater in the 900 mg/day (32%) and 1200 mg/day (33%) groups were both superior to placebo (16%, P = 0.024 for both). The CGI scores for each dose of ezogabine were better than those in the placebo group as well (P = 0.015, 0.004 and 0.005 for the 600 mg, 900 mg and 1200 mg doses respectively).
The RESTORE1 trial (Retigabine Efficacy and Safety Trials for Partial Onset Epilepsy) was a randomized, double-blind, placebo-controlled trial designed to study the use of a 1200 mg/day dose (divided) of retigabine in patients with SP or CP seizures with or without generalization. 27 Subjects (n = 306) were between the ages of 18–75 years, and must have had 4 seizures over the eight weeks preceding the double-blind phase. The titration phase of the trial lasted for six weeks, and was followed by a 12-week maintenance phase. Doses were initiated at 100 mg given three times daily, and were titrated by 150 mg/day at weekly intervals. A single decrease of 50 mg/dose was allowed one time at week 7 if necessary due to intolerance. Subjects were allowed to be on up to three additional antiepileptic medications or a vagus nerve stimulator as long as no changes were made for the duration of the study. Data was collected from patient diaries, and CGI scores were assessed by the investigators. In addition, a similarly-scored scale measuring patient impressions of improvement (PGI) was also completed by participants. The primary endpoint was defined as the percent change in seizure frequency over 28 days, and the secondary end point was the responder rate with a 50% threshold, also over 28 days. The median decrease in seizure frequency was significant in favor of ezogabine (44.3% vs. 17.5%, P < 0.001), as was the responder rate (44.4% vs. 17.8%, P < 0.001) during the entire double-blind phase. Both criteria were also met throughout the maintenance phase (primary end-point: 54.5% vs. 18.9%; responder rate: 55.5% vs. 22.6%, P < 0.001 for both). No difference in seizure frequency was observed between groups remaining seizure free during the double-blind portion of the study, however considering the entire cohort, the median percent of seizure-free days was greater for ezogabine than for placebo throughout (P < 0.001). CGI scores also favored ezogabine for the duration of the study. PGI scores were not different at any time point for ezogabine vs. placebo.
RESTORE 2 was similar to RESTORE 1 with regard to primary and secondary outcomes, though CGI and PGI scores were not measured. 10 Doses of ezogabine utilized in this study for comparison to placebo were the lower doses of 600 mg/day and 900 mg/day. RESTORE 2 had four phases. The first was an eight-week baseline, which was followed by a four-week dose titration phase (100 mg three times daily titrated to target over two-four weeks depending on dose). The maintenance phase was 12 weeks long, and was followed by a transition phase for those patients agreeing to participate in an open-label extension of the original study. As with RESTORE 1, patient diaries were utilized for data collection. Of 471 subjects, significantly more met both the primary endpoints (28-day decrease in seizure frequency during the double blind phase (600 mg dose (27.9%, P = 0.007) and 900 mg dose (39.9%, P < 0.001) vs. placebo (15.9%)) and the responder rate during the maintenance phase (600 mg dose (38.6%) and 900 mg dose (47%) vs. placebo (18.9%), P < 0.001 for both)). Responder rates within a pre-defined completers population during the maintenance phase were also significant (P < 0.001) for subjects taking either of the active treatment doses (39.3%, 49.6% and 19.6% for 600 mg, 900 mg and placebo respectively). A comparison across all groups revealed no difference in the rate of complete freedom from seizures.
Safety and tolerability
The adverse events most often noted in clinical trials were related to the CNS. In addition, rates of adverse events, and by extension study dropouts, were significantly higher during the forced titration phases of the clinical trials than during any maintenance phase. There did not appear to be any correlation with frequency of adverse events and duration of treatment. Furthermore, the majority of observed adverse events were dose-related, and mild to moderate in nature.10,26,27 Adverse events noted in at least 10% of patients across the studies included somnolence, fatigue, confusion, dizziness, tremor, abnormal thinking, vertigo, speech disorders and amnesia. Headache and asthenia were among the most frequent adverse events not of CNS origin. Psychiatric events including hallucinations and depression occurred in a small number of patients. Ezogabine has not been definitively associated with any negative laboratory, electrocardiogram, or vital sign events.
Serious adverse have been rare. In the Porter study, 21 subjects receiving active drug experienced adverse events defined as serious (including isolated cases of suicidal ideation and psychosis) compared with 8 receiving placebo. There were 79 withdrawals, including 17 in the placebo arm. No deaths occurred in this study. 26 Serious adverse events in RESTORE 1 occurred in 19 subjects receiving ezogabine and 8 receiving placebo, and were classified as nervous system disorders (4% ezogabine, 1% placebo), psychiatric disorders (3% ezogabine, 1% placebo) or metabolism/nutritional disorders (2% ezogabine). There was one death considered to be possibly related to treatment in a patient with pre-existing moderate fasting hyperglycemia and cardiovascular disease who developed diabetic ketoacidosis while taking ezogabine. Seventy-one subjects in the active treatment group withdrew from the study compared to 11 in the placebo group. 27 During the double-blind phase of the RESTORE 2 trial, 4% of placebo subjects and 8% of ezogabine subjects reported serious adverse events. A single treatment emergent death occurred in a subject receiving ezogabine. There was also one death in the placebo arm and one in a study subject that had yet to receive a dose of study drug. Convulsions occurred in a single subject taking placebo, and in three subjects in each of the active treatment arms. The percentages of discontinuation due to adverse events were 8% for placebo, 17% for ezogabine 600 mg/day and 26% for ezogabine 900 mg/day. 10
Because K+ channels are present in multiple tissues throughout the body, particular attention has been given to ezogabine and its potential to exert effects outside of the CNS. The drug has not been shown to affect K+ channels located in cardiac tissue (Kv7.1 channels coded by gene KCNQ1), as the glycine target needed for ezogabine binding is absent. 9 However, there is an overlap of K+ channel types located in the CNS and in the urothelium of the bladder. 28 An analysis of 1365 subjects exposed to ezogabine revealed voiding difficulties in 8.6%, with an additional 3% reporting slight increases in post-void residual volume (PVR). Five subjects required urinary catheterization. 29 One subject required continuing self-catheterization after drug discontinuation. Urinary involvement was also evident in the clinical trial data submitted to the FDA. One subject enrolled in the RESTORE 1 trial reported urinary retention while taking ezogabine (compared with two receiving placebo), and 15 (vs. six) demonstrated an increase PVR volume of at least 100 ml. Overall, the study participants reported episodes of urinary tract infection, hesitation, dysuria, and asymptomatic chromaturia (a known side-effect of ezogabine use unrelated to blood in the urine). In general, symptoms improved or resolved either spontaneously or with drug discontinuation. 27 No treatment group in the RESTORE 2 trial experienced any significant changes to American Urological Association Symptom Index scores or PVR. Chromaturia was present in one patient receiving placebo, and three receiving ezogabine. Nephritis or urinary retention resulted in study withdrawal in three subjects. 10
Risk Evaluation and Mitigation Strategy (REMS)
The goal of the REMS for ezogabine is to adequately inform health care providers of the potential for urinary symptoms, and retention in particular. 30 As part of the REMS, a website will be made available via direct link from the Valeant Pharmaceuticals North America website. Data will be required to be submitted to the FDA each of the first three years the drug is marketed, and again at seven years from the date of the original REMS approval.
Potential ezogabine patients should be evaluated for greater likelihood of experiencing urinary retention. Risk factors would presumably include enlarged prostate, anti-cholinergic medication use, and cognitive impairments that might make regular voiding and recognition of symptoms difficult. Though not considered absolute contraindications to use, circumstances such as these warrant a comprehensive pre-therapy evaluation of urinary function and regular monitoring during ezogabine treatment. A medication guide is to be dispensed with all ezogabine prescriptions and samples, and will instruct patients to self-monitor for difficulty initiating urination or emptying the bladder, weak urine stream, and dysuria.
Summary
Ezogabine represents a new class of antiepileptic medication for the adjunctive treatment of refractory partial seizures. Study data have demonstrated clinical efficacy similar to other medications used for this purpose. In general, side-effects and efficacy seem to be best balanced at a dose of 300 mg given three times daily, however, doses up to 1200 mg have been used in clinical trials. 9 Rapid, forced titration of the medication is associated with frequent CNS-related adverse events, though the majority of these events tend to be mild to moderate in nature. Slow titration is likely to result in better drug tolerance and fewer treatment withdrawals. Urinary retention may occur with ezogabine, and patients should be screened for risk factors prior to treatment initiation. In addition, all patients should be counseled about signs of adverse events involving the urinary tract. Marketing of the drug is currently awaiting schedule designation owing to reports of benzodiazepine-like effects. The drug is approved for adults only, and has not been studied in generalized epilepsy.
Disclosures
Author(s) have provided signed confirmations to the publisher of their compliance with all applicable legal and ethical obligations in respect to declaration of conflicts of interest, funding, authorship and contributorship, and compliance with ethical requirements in respect to treatment of human and animal test subjects. If this article contains identifiable human subject(s) author(s) were required to supply signed patient consent prior to publication. Author(s) have confirmed that the published article is unique and not under consideration nor published by any other publication and that they have consent to reproduce any copyrighted material. The peer reviewers declared no conflicts of interest.