
Abstract
Combining opioid agonists with ultra-low (typically pg-ng/kg) doses of the opioid antagonists naloxone or naltrexone can enhance analgesic efficacy and decrease certain side effects. Such combinations have also been shown to decrease tolerance, dependence and addictive potential in animals. We now know that ultra-low-dose naloxone/naltrexone effects occur by preventing a G protein coupling switch (Gi/o to Gs) of the mu opioid receptor (MOR) that occurs briefly after acute administration or persistently after chronic administration of opioids. The picomolar binding site for naloxone or naltrexone in these effects is on the scaffolding protein filamin A (FLNA), rather than on opioid receptors. Interestingly, a second, nanomolar binding site on FLNA may disrupt the benefits of binding the picomolar site, perhaps explaining why ultra-low-dose naloxone/naltrexone effects vanish at just slightly higher doses. A novel analgesic drug candidate aims to avoid this issue by binding only the high-affinity FLNA site, while simultaneously activating MOR. Binding this single site on FLNA also provides anti-inflammatory activity.
Paradoxical Effects of Ultra-Low-Dose Opioid Antagonists
The study of the paradoxical effects of ultra-low-dose naloxone or naltrexone started over 20 years ago, with the initial finding that while opioid agonists normally inhibit, or shorten, the action potential duration of dorsal root ganglion cells, lower doses of opioid agonists induce an opposite, excitatory effect: a prolongation of the action potential.1,2 Crain and Shen subsequently showed that ultra-low-dose naloxone blocked the action potential prolongation at low picomolar concentrations, and in vivo, ultra-low-dose naltrexone (10 ng/kg, s.c.) enhanced and prolonged morphine's antinociceptive potency. 3 This seminal report also included data demonstrating that co-treatment with ultra-low-dose naltrexone dramatically attenuated morphine withdrawal and partially prevented analgesic tolerance.
Since then, many more preclinical studies have shown ultra-low-dose naloxone or naltrexone to enhance and prolong opioid analgesia, to reduce tolerance and dependence,4–10 and even to reverse hyperalgesia caused by acute, low-dose opioids to produce analgesia. 11 Figure 1 illustrates a clear blockade of analgesic tolerance. 5 Later work showed ultra-low-dose naltrexone to attenuate opioid reward or addictive properties in conditioned place preference 12 and self-administration and reinstatement paradigms. 13 These preclinical studies of addictive properties dissociated the analgesic from the addictive effects, since ultra-low-dose naltrexone enhanced one while suppressing the other. Overall, the combination of ultra-low-dose naloxone or naltrexone with opioid agonists can significantly enhance analgesia while diminishing tolerance, dependence and perhaps addictive properties. They continue to be administered together by many physicians with positive results. Nevertheless, clinical studies of ultra-low-dose naloxone or naltrexone combined with opioids have been mixed, with the precise ultra-low dose a critical variable.
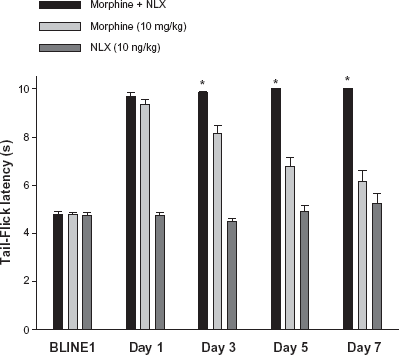
Co-treatment with ultra-low-dose naloxone (10 ng/kg) prevented the antinociceptive tolerance caused by chronic morphine (10 mg/kg, s.c., twice daily for 7 days). Rats treated with morphine + naloxone showed stable tail-flick latencies over the week of treatment, whereas tail-flick latencies of rats receiving morphine alone declined to a level not significantly different from naloxone alone.
P < 0.05 for morphine + NLX versus morphine. Reprinted from, 5 with permission from Elsevier.
Clinical Experience with Ultra-Low-Dose Opioid Antagonists
Clinical experience with opioid antagonists combined with opiates is limited to case reports, small clinical studies,14–18 and two large, well-controlled clinical trials.19,20 In one case study, a diabetic polyneuropathy patient, who previously had no pain relief from a variety of treatments, reported profound analgesia when 2 μg/day of naltrexone was co-administered with methadone. 15 In another case study of severe, chronic low back pain refractory even to 50 mg intrathecal morphine, a daily intrathecal infusion of 5 mg morphine plus 20 ng naloxone reduced pain by up to 80% for the three-year follow-up period. 21 The first controlled clinical study demonstrated an opioid-sparing effect and a reduction in side effects by a continuous infusion of naloxone at 0.25 μg/kg/hr added to morphine administered by patient controlled analgesia (PCA). 14 Using historical controls, similar opioid-sparing results were reported from the same naloxone infusion rate administered to elderly patients receiving tramadol after joint replacement surgery. 22 Using nalmefene, an opioid antagonist not yet confirmed to bind filamin A, another study reported decreased severity of pain 24 hours later and a decreased need for antiemetics and antipruretics following a single 15 or 25 μg injection of nalmefene prior to morphine PCA. 18 Cepeda and colleagues were, however, unable to replicate these effects using a higher dose of naloxone mixed with morphine for PCA 16 and subsequently showed only a decrease in side effects using a much lower naloxone dose. 17 More recently, buprenorphine analgesia was significantly enhanced by 2.3 μg naloxone for a 70 kg patient but not by higher and lower naloxone doses. 23
Although it is difficult to compare doses of naloxone or naltrexone in these small clinical studies, it is clear that the analgesic-enhancing effect is not consistently seen, even if dose levels seem to be consistent. A clear naltrexone dose-effect relationship was noted, however, in two clinical trials of Oxytrex™ (oxycodone + ultra-low-dose naltrexone). The first showed a significant increase in analgesia over oxycodone alone if patients received 2 but not 4 μg/day of naltrexone. 19 Although there was no interaction between treatment and gender, there was a greater treatment effect in men (P = 0.01) than in women (P = 0.056). Nevertheless, a significant difference between Oxytrex and oxycodone occurred in women but not in men, possibly due to the small number of men in the study (Fig. 2). In a phase III trial of Oxytrex, significant decreases in physical dependence as well as in constipation, somnolence and pruritis were observed in patients receiving 2 but not 4 μg/day of naltrexone. 20 In this latter trial, patients were allowed to titrate their dose by taking higher (or lower) strength tablets to achieve sufficient analgesia, but with the naltrexone in Oxytrex tablets fixed at 1 μg/tablet as in the first study, patients received a consistent 2 or 4 μg naltrexone/day, depending on bid or qid dose regimens. Interestingly, the total average daily dose after self-titration was 12% lower for both bid and qid Oxytrex groups compared to the qid group receiving oxycodone with no naltrexone. We believe the bid versus qid differences in both trials are due to the total daily dose of naltrexone and represent a crossing of the effective “ultra-low-dose” threshold.
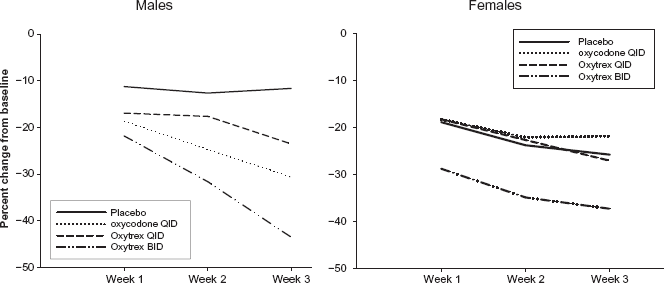
Reduction in pain intensity in males and females by Oxytrex. Oxytrex BID, a daily dose of 2 ug naltrexone, provided the greatest reduction in pain intensity scores in both males and females. At Week 3, Oxytrex BID was significantly better than placebo in males and significantly better than oxycodone in females. The pain intensity scores of the Oxytrex QID group, receiving 4 ug naltrexone/day, did not statistically separate from placebo. Reprinted from 19 with permission from Elsevier.
The only clinical study to assess addictive potential of oxycodone with and without ultra-low-dose naltrexone was a double-blind, placebo-controlled, cross-over study of 14 experienced opioid abusers. 24 Although the study detected differences between oxycodone and placebo and between 20 and 40 mg of oxycodone on positive subjective ratings, adding 1 or 0.1 mg naltrexone to either dose of oxycodone produced no significant differences. This result is at odds with preclinical data showing reduced addictive potential from the addition of ultra-low-dose naltrexone.12,13
Variables in Defining an “Ultra-Low” Dose
Why was ultra-low-dose naltrexone or naloxone not consistently effective? The most significant variable, as noted in the Phase III clinical trial mentioned above, was the precise, ultra-low dose. Ultra-low-dose naloxone and naltrexone effects do not follow a typical dose-response pattern, but usually span several log units in the pg/kg to ng/kg range. There is a clear threshold, however, where increasing the dose of naltrexone or naloxone starts to disrupt their effects at ultra-low doses. Why this dose threshold is so crucial will become clearer in later sections discussing the mechanism of action and filamin A binding site. Unfortunately also, this fine line seems to shift with sex, strain, age, nociceptive versus neuropathic pain, and possibly many other variables.
The following two studies show the optimal doses of naltrexone can vary with the opioid agonist it is combined with as well as the sex of the animal. The prevention of tolerance and withdrawal-associated hyperalgesia by ultra-low-dose naltrexone was demonstrated at 0.3, 3, 300 and 3000 ng/kg naltrexone when combined with 3 mg/kg morphine 10 and at 1 pg/kg or 1 ng/kg naltrexone when combined with oxycodone. 9 The higher naltrexone doses used in these studies were used in males. Hence, although male rodents are generally more sensitive to opioid analgesia, 25 they appear to be less sensitive to ultra-low-dose naltrexone. These findings likely contribute to the variations in optimal ultra-low-dose naltrexone dose ranges noted both with sex of the animal and the opioid used in combination. Why the choice in opioid agonist matters may be related to different rates in effecting G protein coupling, since naloxone or naltrexone work by preventing a switch in G protein coupling, as discussed below.
In addition to sex differences, the effects of ultra-low-dose naltrexone on morphine antinociception and tolerance can also vary with rat strain. 26 Whereas the enhancement of morphine antinociception and attenuation of morphine tolerance was observed in both Sprague-Dawley and Long-Evans rats, naltrexone (0.1–100 ng/kg) did not enhance morphine antinociception in Fisher 344 or Lewis rats, nor did 10 or 100 ng/kg naltrexone significantly attenuate tolerance in these two strains. Given that pg/kg doses of naltrexone most effectively enhanced oxycodone antinociception in female mice, 9 perhaps this even lower dose range might enhance antinociception in these two rat strains. Terner (2006) also reported strain-dependent sex differences. Another example of strain differences is the initial report that ultra-low-dose naltrexone enhances morphine antinociception in Swiss Webster mice but antagonizes it in 129/SvEv mice. 27 A final study in rats noted sex effects as well as age effects: morphine antinociception was enhanced by low-dose naltrexone in mature female but not mature male rats (18–22 weeks) and was negligible in younger rats. 28 However, the naltrexone dose range used in that study was again comparatively high (0.002–2 μg/kg), and the effect in mature females was “inversely related to dose”.
Neuropathic pain is typically not effectively treated by opioids, but ultra-low-dose naltrexone can enhance their effects. Interestingly, the doses of naltrexone used to enhance the anti-hyperalgesic effects of oxycodone are notably higher than the dose ranges mentioned above for nociceptive pain. In the spinal nerve ligation model of neuropathic pain, naltrexone enhanced the effectiveness of oxycodone and greatly diminished the tolerance seen with repeated administrations. 29 Naltrexone was administered at 3 μg/kg, a dose 10- to 100-fold higher than those used to enhance analgesia and alleviate tolerance in nociceptive pain.
In summary, all of these studies show that an effective ultra-low dose of naloxone or naltrexone seems to depend on many interacting variables, including sex, strain, age, the opioid agonist used in combination, and type of pain. This variability or inconsistency in the effective dose range made little sense prior to the discovery that FLNA is the target protein for naloxone and naltrexone in enhancing opioid analgesia. As discussed below, FLNA is a large, dynamic scaffolding protein interacting with many different receptors and intracellular proteins. These variables of sex and strain, etc., that appear to influence the effective ultra-low dose may reflect underlying differences in FLNA associations, conformations and/or content. More critical however, is the fact that naloxone and naltrexone bind this large protein at two different sites, and with a 200-fold difference in affinity.
Molecular Mechanism: Preventing a G Protein Coupling Switch
Long before discovering the target or even the consequences of its binding, the mechanism of action of ultra-low-dose naloxone or naltrexone was hypothesized as a blockade of excitatory signaling opioid receptors.3–5,30 Ten years later, we demonstrated that the excitatory signaling of opioid receptors was due to their coupling to Gs proteins as earlier hypothesized, with one important distinction. Ultra-low-dose opioid antagonists were initially thought to bind preferentially a subset of mu opioid receptors (MOR)30,31 that couple to Gs instead of the typical coupling to Gi and Go proteins that subsequently inhibit adenylyl cyclase. 32 (The activated Gs proteins would instead stimulate adenylyl cyclase and downstream cellular activities.) Our data suggested that instead of binding to a discreet population of receptors, ultra-low-dose naloxone or naltrexone controlled the coupling preference of the MOR. Specifically, we showed that co-treatment with 10 ng/kg naloxone prevented a chronic morphine-induced, Gi/o-to-Gs switch in G protein coupling by the MOR as well as a coincident interaction of the Gβγ dimer with adenylyl cyclase II and I V. 5 Chronic morphine treatment both diminishes the native coupling to Gi/o and instates a novel coupling to Gs proteins by MOR.5,31 We subsequently confirmed that the Gβγ interacting with adenylyl cyclases originates from the Gs protein coupling to MOR 33 and not from MOR's native G proteins as earlier postulated. 34
The molecular pharmacology underlying ultra-low-dose naloxone's or naltrexone's prevention of opioid tolerance and dependence had been demonstrated ten years after the initial in vivo demonstration. It was clear that ultra-low-dose naloxone blocked the MOR–-Gs coupling associated with chronic opioid administration, while simultaneously enhancing levels of coupling to MOR's native G proteins. Strikingly, our co-immunoprecipitation data showed that in spinal cord of rats co-treated with morphine plus ultra-low-dose naloxone, MOR–-Gi/o coupling levels greatly surpassed those of opioid-naïve rats (Fig. 3), 5 perhaps indicating the enhanced analgesia seen acutely with these combinations. Despite this elucidation of mechanism of action of ultra-low-dose naloxone or naltrexone, their actual binding site in regulating MOR coupling was unknown.
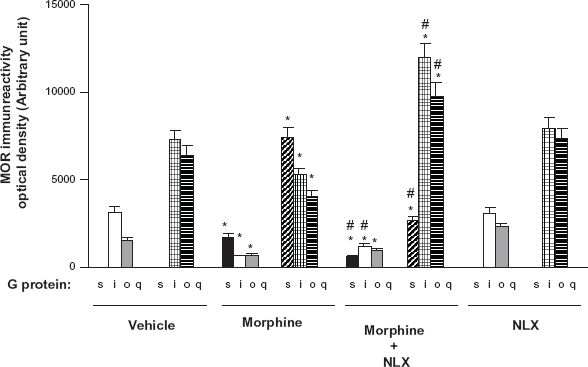
Ultra-low-dose naloxone blocks chronic morphine-induced MOR–-Gs coupling and dramatically enhances MOR–-Gi/o coupling. Densitometric quantitation of western blot of co-immunoprecipitation with Gα proteins and MOR in spinal cord tissue of rats treated twice daily for 7 days with 10 mg/kg morphine, 10 ng/kg naloxone, or the combination. Data are means S.E.M. derived from four individual rats in each treatment group. Solid bars indicate basal coupling, and hatched bars indicate coupling after receptor stimulation by in vitro morphine.
Finding the Target
Because naloxone prevents MOR–-Gs coupling at concentrations well below its affinity for MOR and by influencing the coupling behavior of MORs, we considered proteins that interact with MOR and MOR-associated G proteins as the most likely targets, particularly those able to interact with multiple MORs. We first examined proteins that co-immunoprecipitated with MOR during activation. We identified a 300-kDa protein co-immunoprecipitating with MOR as filamin A (FLNA, 3CNK.pdb) and then demonstrated that naloxone and naltrexone bind to FLNA with 4 pM affinity. 35 This high-affinity interaction certainly could mediate the effects of ultra-low doses of these two compounds.
Best known for cross-linking cytoplasmic actin into dynamic scaffolds to control cell motility, filamins are large cytoplasmic proteins increasingly found to regulate cell signaling by interacting with over 30 different receptors and signaling molecules,36,37 including MOR. 38 We deduced the precise, pentapeptide binding site on FLNA by using overlapping peptides within the C-terminal, since C-terminal FLNA was earlier shown to interact with MOR using a yeast-two hybrid. 38 We demonstrated the functional significance of this high-affinity interaction by using peptide fragments containing the binding site to prevent naloxone from binding full-length FLNA in organotypic striatal slice cultures. Without addition of the peptide, FLNA interacts with ultra-low-dose naloxone and naltrexone to prevent chronic morphine-induced MOR–-Gs coupling, but in the presence of the peptide fragment, naloxone loses its ability to block the Gs coupling (Fig. 4).
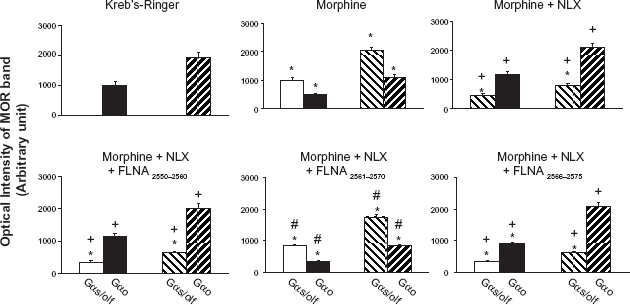
FLNA2561–2565 blocks 10 pM naloxone's prevention of the chronic morphine-induced Go-to-Gs coupling switch. Striatal slices were chronically treated with vehicle, morphine, naloxone (NLX), morphine + NLX, or with one of the three FLNA peptides alone or in combination with morphine + NLX. Coupling between MOR and Gs/Go proteins was assessed by Western blot and analyzed by densitometric scanning. Chronic morphine exposure caused a Go-to-Gs coupling switch that was blocked by NLX co-treatment. NLX's suppression of this coupling switch was blocked by FLNA2561–2570 but not by FLNA2566–2575 or FLNA2550–2560, illustrating that NLX's protective effect occurs via its binding to FLNA within FLNA2561–2570. Solid bars indicate basal coupling; hatched bars indicate coupling after in vitro morphine stimulation. n = 6.
This high-affinity binding site in C-terminal FLNA therefore appears to underlie the paradoxical enhancement of opioid analgesia and prevention of analgesic tolerance and dependence by ultra-low-dose opioid antagonists. The high-affinity interaction of naloxone or naltrexone with FLNA likely prevents a critical MOR–-FLNA interaction that otherwise increases the probability of MOR coupling to Gs instead of Gi/o proteins. In identifying the binding site though which ultra-low-dose naloxone or naltrexone prevent MOR–-Gs coupling, our data also revealed an important regulation of MOR–-G protein coupling by filamin A. Additionally, the discovery of a novel, non-opioid binding site of naloxone and naltrexone underlying their “ultra-low-dose” effects opens avenues to developing a novel class of analgesics without the difficulties and constraints of combining ultra-low-dose opioid antagonists with opioid agonists.
Two Binding Sites on Filamin A
One wonders how it can be that 2 but not 4 μg/day of naltrexone can enhance analgesia clinically, and how such a broad dose range below this threshold can be effective while crossing the line, as variable as it is, destroys the effects. Intuitively, the obvious thought was that the additional opioid antagonist starts to disrupt the opioid agonist, thereby diminishing the overall effect. However, a nanogram or microgram dose of opioid antagonist is not likely to noticeably disrupt the biological effects of a milligram dose of opioid agonist. Evidence that the possibility of spillover to opioid receptors is not the case comes from a study examining the reduction in rewarding properties of oxycodone by ultra-low dose naltrexone. 12 In this study, 0.03 or 0.3 ng/kg naltrexone blocked a conditioned place preference (CPP) to 3 mg/kg oxycodone, whereas 3 ng/kg naltrexone did not block this CPP to oxycodone (Fig. 5). As the naltrexone dose increased to 30 ng/kg, the CPP to oxycodone is not significant. The robust CPP in the middle of this dose-response curve demonstrates a point where the “ultra-low-dose effects” are lost and the classical antagonism of opioid receptors is not yet occurring. In analgesic paradigms, if opioid receptors are antagonized, analgesia would be decreased, just as it would when crossing the ultra-low-dose threshold. However, in the CPP paradigm, when the ultra-low-dose effect is lost, the CPP to oxycodone reappears; whereas, antagonizing opioid receptors would interfere with the CPP.
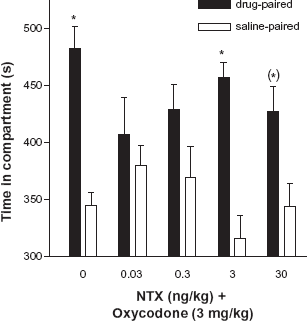
Dose-response of naltrexone on a CPP to oxycodone. Bars represent the mean (± SEM) amount of time (s) spent in saline- and drug-paired compartments on test day. Oxycodone (3 mg/kg, s.c.) produced a significant CPP that was blocked by the addition of naltrexone (NTX) at 0.03 ng/kg s.c., or 0.3 ng/kg. In contrast, NTX at 3 ng/kg s.c. did not block the CPP to oxycodone. Oxycodone combined with the highest dose of NTX, 30 ng/kg s.c., produced only a trend toward a CPP. Reprinted from 12 with permission from Springer-Verlag.
What then is disrupting the ultra-low-dose naloxone/naltrexone effects, making this threshold of efficacy such a fine line to tread? We believe this threshold is due to a second binding site of naloxone and naltrexone on the FLNA protein. A competition (displacement) curve for the inhibition of tritiated naloxone binding by naltrexone to membranes from FLNA-expressing cells shows two affinity states with IC50-high of 3.94 picomolar and IC50-low of 834 picomolar (Fig. 6). 35 It is very possible that binding the almost nanomolar binding site on FLNA may alter the conformation of the protein or disrupt some conformational change induced by binding the picomolar binding site. This 200-fold difference in affinity could represent the fine line of whether ultra-low-dose naloxone or naltrexone enhances opioid analagesia, prevents tolerance and dependence and attenuates opioid reward or not. To avoid this problem, we simply had to avoid the second binding site on FLNA. Hence, we started the search for a novel compound that activates opioid receptors and simultaneously binds the high-affinity site on FLNA with picomolar or better affinity.
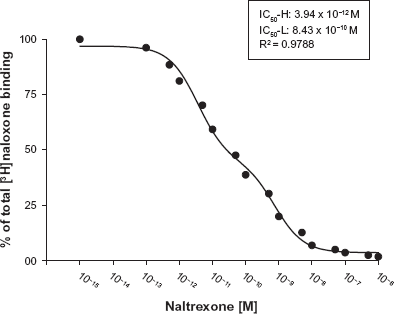
Naloxone binds FLNA with picomolar affinity. A competition (displacement) curve for the inhibition of [3H]-naloxone binding by naltrexone to membranes from FLNA-expressing A7 cells shows two affinity states with IC50-H of 3.94 picomolar and IC50-L of 834 picomolar. A nonlinear curve-fit analysis was performed using a competition equation that assumed two saturable sites for the naltrexone curve comprising of 16 concentrations ranging from 0.1 pM to 1 μM. Data are derived from 6 experiments each using a different set of A7 cells. Reprinted from. 35
A New Approach with Novel Compounds
Once the high-affinity binding site of naloxone and naltrexone was found to be a pentapeptide region on FLNA, we were able to design novel compounds that combine high-affinity FLNA binding with MOR agonist activity in a single, new chemical entity. Our approach was molecular modeling, virtual screening of the world's compounds against the model, followed by actual screening of a small number of selected compounds for binding to the FLNA pentapeptide and activation of MOR. The screening data was used to refine the model, and the cycle was repeated once more before initiating medicinal chemistry, which produced several good lead candidates with fM to pM affinity to the FLNA pentapeptide and good analgesic efficacy in mice by oral administration. These novel compounds do not structurally resemble known opioids, and interestingly, the three lead compounds that were tested do not displace diprenorphine in a binding assay, suggesting a novel site in activating MOR. Their dual function avoids the difficulties of developing a combination product, allowing easier formulation and simpler clinical trials. Most importantly, by screening for compounds that bind only the picomolar binding site on FLNA and not the second, lower affinity site on this scaffolding protein, we avoid the difficulties in treading the ever-moving, fine line of the ultra-low dose of naloxone or naltrexone. Further, greater efficacy is expected since these picomolar binding sites can be fully saturated. To confirm that this sand-trap is now a non-issue, we can simply increase concentrations of these compounds and confirm that effects are not lost. What is expected is an analgesic suited for moderate-to-severe pain with little to no tolerance, dependence and addictive potential. We also anticipated some anti-inflammatory activity, given extensive prior research showing that (+) naloxone, which binds FLNA but not opioid receptors, suppresses glial activation, and at femtomolar concentrations.39–41
To confirm that our compounds do not binding the lower-affinity site on FLNA and that they maintain activity as concentrations increase, we tested increasing doses in an in vitro anti-inflammatory test. Previous work with (+)naloxone showed that its anti-inflammatory effect is lost in the high picomolar range before reappearing in the micromolar range.39,40 This threshold is reminiscent of the loss of effects of naloxone and naltrexone in potentiating opioid analgesia, and knowing that naltrexone binds FLNA in the femtomolar to picomolar range, we suspected that these effects could be the result of FLNA binding. The target suggested by the Watkins laboratory in these effects is toll-like receptor 4 (TLR4) of innate immune cells, because naloxone and naltrexone, including their isomers that are inactive at opioid receptors, block TLR4 signaling.41,42 A large body of work, led by Dr. Watkins, has demonstrated the role of glial activation in the negative effects of opioids, including opioid tolerance, dependence, reward, and respiratory depression, 43 so it is possible that ultra-low-dose opioid antagonists are alleviating these effects by diminishing the glial activation of opioids. Two other laboratories also demonstrated that ultra-low-dose naloxone or naltrexone attenuates glial activation in spinal cord associated with morphine analgesic tolerance.44,45 Although the glial activation could be a downstream effect of MOR–-Gs signaling, the cell line used to demonstrate that (+) and (-) naloxone disrupt TLR4 signaling was devoid of opioid receptors. 42 Hence, binding to FLNA in glial cells may slow down vesicular release of cytokines, or disrupt TLR4 signaling.
We first assessed cytokine release from LPS-stimulated human astrocytes and the potential blockade of this cytokine release by our current lead, named PTI-609, and two related novel compounds. We assessed concentrations from 100 fM to 100 nM and compared these to 10 pM and 1 nM of (+)naloxone. Whereas all three of our compounds potently reduced release of IL-6, IL-1β and TNF-α, (+)naloxone suppressed release of these cytokines somewhat less potently at 10 pM and virtually not at all at 1 nM (Fig. 7). This loss of efficacy by (+)naloxone at 1 nM again demonstrates the crossing of the ultra-low-dose threshold, likely mediated by binding to the second binding site on FLNA, the site it binds with 834 pM affinity. We also tested PTI-609 administered orally twice daily in the rat collagen-induced arthritis model. The PTI-609 analog significantly reduced foot volume at 56 mg/kg. The reduction in foot volume was much less than that produced by 3 mg/kg celecoxib and approximately half that seen with 25 mg/kg ibuprofen in this initial proof of concept study.
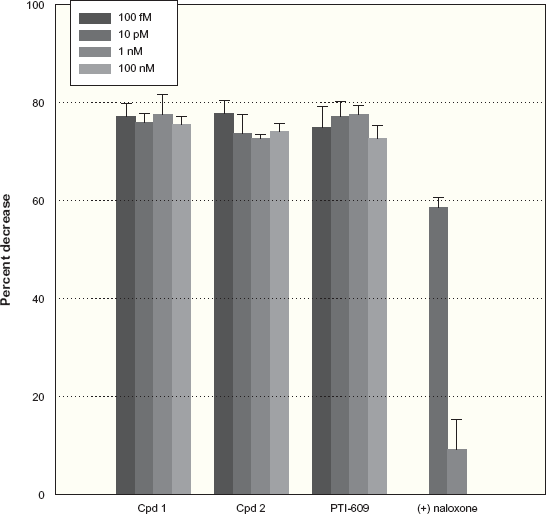
FLNA-binding compounds block LPS-stimulated cytokine release from human astrocytes. Three FLNA-binding compounds block TNF-α release from primary cultures of human astrocytes by 75% or more at concentrations ranging from 100 fM to 100 nM. (+) Naloxone blocked TNF-α release by almost 60% at 10 pM, but this suppression was nearly abolished at 1 nM. Similar results were obtained with IL-1β and IL-6. Data are means ± SEM. Reprinted from 55 with permission.
Addictive Potential
Although one clinical study failed to show a decrease in addictive potential with ultra-low-dose naltrexone combined with oxycodone, we believe that since PTI-609 is not hampered by binding to the second FLNA binding site, the preclinical data might have a better chance of translating to a reduced addictive potential in humans. Our data linking the immediate and transient MOR–-Gs coupling to CREB activation, as well as the blockade of conditioned place preference (CPP) by pg/kg doses of naltrexone, suggested that our novel compounds would have minimal addictive potential. We previously showed that in brain slices, acute high dose morphine caused an immediate but transient Gs coupling by MOR that was associated with activation of cyclic AMP response element-binding protein (CREB), 46 a transcription factor implicated in addiction and withdrawal effects.47–51 Blockade of this immediate MOR–-Gs coupling by picomolar concentrations of naloxone or by its pentapeptide binding site on FLNA blocked the associated activation of CREB. The rewarding effects of opioid drugs are most pronounced as plasma levels are rising quickly and are less intense or even negative later,52–54 suggesting that this immediate Gs coupling is at least temporally associated with the most intense phase of opioid reward. To test the hypothesis that our FLNA-binding novel compounds would not have the same rewarding properties of classical opioids, we examined equi-analgesic doses of PTI-609 and oxycodone in the CPP paradigm. Oral doses that produced an 80%–90% analgesic effect as well as a dose ¼ log lower were compared to a vehicle group, using three conditioning sessions to the drug-paired compartment as well as to the vehicle-paired compartment. A significant CPP to both doses of oxycodone was observed, whereas in contrast, there was no change from baseline in time spent in the putative conditioning side with either dose of PTI-609 (Fig. 8). This result suggests that at therapeutic doses, no rewarding effects occur, or at least none strong enough to produce a CPP. We attribute this lack of CPP to the FLNA-binding capacity of PTI-609 and its prevention of MOR–-Gs coupling; however, the activation of MOR via a novel binding domain within MOR, ie, the compound's inability to displace diprenorphine in a binding assay, may also contribute. Additionally, according to the work linking glial activation with opioid reward, 43 the fact that these compounds potently suppress cytokine release by activated glia may also help prevent rewarding effects despite activation of MOR.
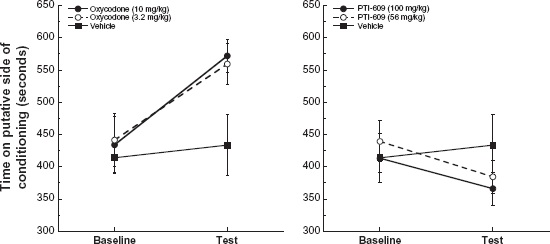
Lack of conditioned place preference with PTI-609. In mice given three 20-minute conditioning sessions to oral administration of drug or vehicle versus saline, there was a significant place preference to both doses of oxycodone but no change in time spent in the putative conditioning compartment in the groups receiving PTI-609. Data are means ± SEM. Reprinted from 55 with permission.
Conclusion
Although early preclinical work and many clinical studies combining opioids with ultra-low-dose naloxone or naltrexone showed great promise, the benefits of such combinations proved to be somewhat inconsistent and highly dependent on a dose threshold that shifts with many variables. Now that the high-affinity biniding site of naloxone and naltrexone is known to be on FLNA, which harbors a second, lower-affinity binding site for naloxone and naltrexone, we surmise that the inconsistent effects and difficulties in treading the fine line of the ultra-low-dose were likely due to interference from this second FLNA binding site. Supportive evidence comes from PTI-609, a new chemical entity that, in a single molecule, binds the pentapeptide region of FLNA with high affinity, avoids the problematic second binding site, and activates MOR via a novel binding site. The early research presented here shows PTI-609 and related compounds to be promising analgesic agents that possess additional anti-inflammatory properties that do not vanish with increasing dose and do not carry the abuse and addictive potential of classical opioid drugs. The problems with current opioid painkillers are well-known, and range from acute side effects to tolerance, dependence, abuse, diversion and addiction. These drawbacks demonstrate a great need for a better class of analgesic agents. PTI-609 is a novel anti-inflammatory analgesic that is poised to fulfill that need and meet the goals initially set out by combining opioids with ultra-low-dose naloxone or naltrexone.
Disclosure
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. L.H. Burns is an employee of Pain Therapeutics Inc., the company that funded the development work on PTI-609 as well as earlier work on Oxytrex. The peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.