
Abstract
Immune thrombocytopenic purpura (ITP) is an autoimmune disease characterized by the production of antibodies to circulating platelets. Traditionally it has been regarded as a disorder of increased platelet destruction, so therapies have targeted this mechanism, including corticosteroids, IVIG, splenectomy and rituximab. More recently, it has become clear that decreased platelet production importantly contributes to the thrombocytopenia. The isolation of thrombopoietin (TPO), the ligand for the mpl receptor and the major hormone regulating platelet production, has shed new light on platelet homeostasis and facilitated the development of new therapeutic approaches. First generation TPO-mimetics were removed from clinical trials when some healthy volunteers and patients with cancer developed antibodies against these drugs that cross-reacted with native TPO, causing paradoxical thrombocytopenia. Two second generation TPO-mimetics, romiplostim and eltrombopag, share no structural homology with endogenous TPO and are being studied in a variety of thrombocytopenic states. Romiplostim was approved by the US FDA in August 2008 for the treatment of patients with ITP who had failed at least one prior therapy. This was based on randomized, placebo-controlled phase III studies that showed increased platelet counts in more than 80% of treated patients, with a durable platelet response by stringent criteria in 38% of splenectomized and 61% of non-splenectomized patients compared to 0%–5% in patients receiving placebo. Being non-immunosuppressive, the toxicity profile is favorable, although there are concerns with this class of drugs with thrombotic risks, “rebound thrombocytopenia,” marrow reticulin fibrosis, or even hematopoietic clonal proliferation. Romiplostim may gain an increasing niche in the treatment of ITP.
Introduction
The existence of thrombopoietin (TPO), the major hormone governing platelet production, was suggested by animal experiments almost 50 years ago. 1 In 1994, TPO was finally isolated and cloned almost simultaneously by five different groups.2–6 Native TPO and a truncated, pegylated analog quickly entered clinical trials for thrombocytopenic states, such as those occurring after cancer chemotherapy. Development came to an abrupt halt when some study subjects produced antibodies to these hormones which cross-reacted with native TPO and caused long-lasting thrombocytopenia. 7 Subsequently, second generation TPO-mimetics have been developed.
Romiplostim (formerly AMG-531), a peptide TPO-mimetic, is the first of the second generation drugs to be extensively studied and to reach the market. The current approved indication is for chronic immune thrombocytopenic purpura (ITP). Accumulating data over decades has made it clear that ITP is not only a disorder of increased platelet destruction, but also a problem with blunted platelet production.8–10 One strong confirmatory piece of evidence for this concept is the high response rate to TPO-mimetic therapy in ITP.11,12 Romiplostim may be carving an increasing niche for the treatment of this disease.
Immune Thrombocytopenic Purpura: Classic Understanding of Pathophysiology and Treatment
ITP is an autoimmune disease characterized by the production of antibodies against platelet membrane glycoproteins, which leads to the premature clearance of platelets by phagocytic reticuloendothelial cells. In the past, the abbreviation ITP was used for idiopathic thrombocytopenic purpura, but current awareness of the immune nature of the disease has led to revision of terminology so now the acronym indicates immune thrombocytopenia. 13 The estimated incidence of ITP is 3.3 per 10,000 population/year in adults, and from 2.2 to 5.3 per 10,000/year in children. 14 It has been thought that the incidence of ITP is highest among young women, but there is some evidence of an increased incidence with age and no gender difference in older patients. 15
The clinical course of ITP is significantly different between children and adults. 16 In children, ITP is usually an acute, self-limited disorder resolving within 6 months in 80%–85% of the cases. 17 In contrast, ITP in adults more often has a chronic course with spontaneous remissions much less frequent (on the order of 10%).18,19 Serious morbidity and mortality associated with childhood ITP is very low, and the decision of when or how to treat engenders some controversy. Therapeutic options include observation only, corticosteroids, IVIG or anti-RhD, and studies are beginning to address possible roles for rituximab or TPO-mimetic agents. The childhood variant of ITP will not be further discussed in this review.
ITP can be classified as “primary” or “secondary” depending on whether there is an underlying disease associated with platelet autoantibodies, such as systemic lupus erythematous, hepatitis C virus, human immunodeficiency virus, Helicobacter pylori, antiphospholipid antibody syndrome, lymphoproliferative disease, or pathogenetic drug exposure. 13 The distinction between primary and secondary ITP is at times clinically relevant because of some differences in natural history and treatment choices.
The most common clinical manifestations of ITP are skin and mucous membrane bleeding such as petechiae and purpura. Commonly, the disease is clinically silent, revealed only by the laboratory finding of a low platelet count. The most feared complication of ITP is intracranial hemorrhage. In a retrospective analysis from 17 adult case studies, the risk of fatal hemorrhage was estimated to be between 0.0162 and 0.0389 cases per patient-year at risk (the time at risk defined as the time during which the platelet count was less than 30 x 10 9 /L). 20 Age adjusted rates were less than 0.4% for those younger than 40 years old, 1.2% for the 40–60 age group and 13% for those over 60. Mortality in ITP is due almost entirely to intracranial hemorrhage and to infectious complications related to therapy. 21 This fact strengthens the caveat to be circumspect in deciding who, when and how to treat.
The diagnosis of ITP is one of exclusion. The disorder classically presents as a very low platelet count with maintained red and white cell counts and normal cell morphology on blood smear. Currently available assays for platelet-associated antibodies lack the specificity and sensitivity to be clinically useful.
Not all patients with a diagnosis of ITP should be treated. Currently available therapeutic modalities have real and potential toxicities, so it is prudent to withhold treatment from asymptomatic patients with milder degrees of thrombocytopenia and a low risk of bleeding. Treatment is rarely indicated in patients with platelet counts above 30–50 x 10 9 /L in the absence of other comorbidities such as bleeding due to platelet dysfunction or another hemostatic defect, trauma, surgery, mandated anticoagulation therapy, or in persons whose profession or lifestyle predisposes them to trauma. 21
Until very recently, treatment options focused on counteracting the increased platelet destruction as this was thought to be the major mechanism of the disease. Glucocorticoids continue to be the mainstay of initial treatment. They have been thought to act by paralyzing the phagocytic reticuloendothelial cells, impairing the binding of auto-antibodies to platelets, and, less reliably, diminishing the synthesis of anti-platelet antibodies. They have also been shown to increase marrow platelet production by undetermined mechanisms. 22 Platelet improvement is noted within one to two weeks of starting treatment in 65%–95% of patients. However, platelet counts fall back down in 90% of these responders as steroids are tapered to potentially tolerable levels for long-term use. 23
Patients who fail steroid treatment, either because of no initial response or relapse as the dose is tapered, traditionally underwent splenectomy. The spleen is the primary site for platelet clearance and also a major site for anti-platelet antibody production. Sustained complete responses after splenectomy occur in 60%–80% of patients. 24 In experienced hands, the operative mortality is low (<1%), and perioperative morbidity has been reduced by laparoscopic techniques. Late post-splenectomy sepsis occurs in less than 1% and can be minimized by vaccinations and vigilance. 25
Intravenous immunoglobulin (IVIG) has a transient but rapid effect and is indicated mainly in severe ITP where an immediate platelet response is judged important. Miscellaneous other indications include pregnant patients and pre-operative situations. It is effective in elevating the platelet count in approximately 85% of patients, with 65% achieving normal counts. 26 Some of the postulated mechanisms of action include Fc receptor blockade of reticuloendothelial cells, anti-idiotype effects on auto reactive B cells, and accelerated autoantibody clearance. 26 Intravenous anti-Rho(D) is an alternative to IVIG in the 80% of patients who have Rh positive blood type and who have not been splenectomized, and may also block the reticuloendothelial clearance of platelets. 27 The main drawback of both therapies is the transient nature of responses, typically lasting about three weeks, and they are expensive. IVIG is associated with infusional reactions, headache, aseptic meningitis, thrombotic and renal toxicities, while anti-RhD may cause severe intravascular hemolysis.28,29
Rituximab, an anti CD-20 monoclonal antibody, has gained popularity as a second line therapy for ITP. Overall responses are seen in 53% to 69% of patients, complete responses in 33% to 54%, usually beginning 2–3 weeks after therapy initiation.30,31 Long-lasting remissions are achieved in about one third of patients. 32 The mechanism of action likely involves depletion of auto-reactive B-lymphocytes. Infusion-related reactions are common only with the first course and are generally quite manageable. Infectious complications have been uncommon, but rare side-effects include late-onset neutropenia, progressive multifocal leukoencephalopathy and severe pulmonary reactions.33–35
Danazol, an attenuated androgen, has been used in ITP for more than 25 years. Reported results vary, but generally a good response can be expected in 50%–67% of patients.36–38 Responses are generally durable, although half the time require indefinite maintenance. Most patients tolerate the medication well, and costs are modest. The mechanism of action of danazol is unclear, but one intriguing possibility is that it increases TPO production. 36
Immunosuppressive and cytotoxic therapies are less often used in recent years, mainly as salvage therapies in severely affected and highly refractory cases. Such therapies have included cyclophosphamide, azathioprine and vincristine. In the few patients where immunosuppression is called for; newer agents such as cyclosporine A and mycophenolate mofetil have had increased use due to perceived lower toxicity. Campath-1H, a humanized monoclonal antibody against the CD52 molecule expressed on both B and T lymphocytes, has shown efficacy, but side-effects include a high risk of opportunistic infection. 39 Plasma exchange can be used in desperate situations but is not usually effective.
Thrombopoietin and First Generation TPO-mimetics
Thrombopoietin (TPO) is the primary growth factor regulating platelet production. 1 The existence of TPO was first postulated in 1958 40 and its receptor, c-mpl was cloned 30 years later. 41 In vitro and animal studies showed the importance of c-mpl activation for platelet production but its ligand remained unknown until virtually simultaneous isolation in five different laboratories in 1994.2–6,42,43 TPO is a molecule of 95 kDa (333 amino acids) that shares 50% homology with erythropoietin.44,45 TPO binding to c-mpl stimulates megakaryocyte proliferation and maturation via activation of signaling pathways that result in the rapid tyrosine phosphorylation of mpl, Janus Kinase 2 (JAK2), and signal transducer and activators of transcription (STAT)-3 and STAT-5, which results in the promotion of cell proliferation. 1
TPO is produced predominantly by the liver, although some mRNA is detectable in kidney, bone marrow, spleen, testis, muscle and brain.2,5,6 Very low TPO levels accompany thrombocytopenia in patients with hepatic failure. 46 After liver transplantation, TPO levels and platelet counts rapidly normalize. 47 TPO elaboration is constitutive; that is, the transcription, translation and release of the TPO molecule are constant, independent of feedback mechanisms.46,48 TPO clearance is accomplished mainly by receptor-mediated endocytosis. Hence, it is the platelet and megakaryocyte mass that determine TPO clearance and blood levels of TPO. 49 When platelet counts fall, the clearance of TPO decreases, circulating TPO levels rise, and megakaryocytes are stimulated to proliferate and differentiate. When platelet counts increase, TPO levels are lowered by enhanced clearance, and megakaryocyte activity is blunted.
Soon after the cloning of TPO, the recombinant native protein (rHu-TPO) and a pegylated truncated analog, recombinant human megakaryocyte growth and development factor (PEG-rHuMGDF), entered clinical trials mainly for thrombocytopenias from chemotherapy for solid tumors,50,51 with stem cell transplantation, 52 and in leukemia. 53 However, PEG-rHuMGDF was found to cause paradoxical persistent thrombocytopenia in 13 of 325 healthy volunteers (4%) and 4 of 650 cancer patients (<1%) after the administration of one or more doses. 7 A detailed analysis of 3 of these cases revealed the production of neutralizing IgG antibodies directed against the drug that cross-reacted with endogenous TPO and neutralized its biological activity. This could result in severe, long-lasting thrombocytopenia. When this problem was appreciated, both PEG-rHuMGDF and rHu-TPO were quickly withdrawn from clinical studies and further development.
This setback stimulated a search for second generation TPO-mimetics that could activate the TPO receptor but would share no structural homology with native TPO. Three general classes of second generation TPOs were developed: TPO peptide mimetics, TPO non-peptide mimetics, and TPO antibody mimetics. One TPO peptide mimetic (romiplostim) and one non-peptide mimetic (eltrombopag) have completed phase III clinical trials for ITP, have been approved by the U.S. FDA (in August 2008 and November 2008 respectively), and have reached the market.
Rationale of TPO-mimetics in ITP
Traditionally, ITP was believed to be a disease of increased platelet destruction, a concept dramatically advanced by Harrington's self-experimentation with infusion of ITP plasma. 54 However, a number of lines of evidence accumulated over several decades implicate an important contribution of impaired platelet production.55–57 In one prospective study of platelet survival and turnover in 38 ITP patients, 30% had decreased platelet production, 43% had normal platelet production, and only 27% had increased platelet production. 58 One explanation for decreased platelet production is that anti-platelet antibodies bind to megakaryocytes, inducing apoptosis.8–10 Another reason for a lack of a compensatory increase in platelet production is that TPO levels are lower than expected for the degree of thrombocytopenia in patients with ITP.59–61 TPO levels are elevated in conditions in which there is decreased megakaryocyte mass, such as aplastic anemia, lymphoid malignancies, amega-karyocytic thrombocytopenia, and patients thrombocytopenic after chemotherapy.42,62,63 With ITP, TPO can be cleared by the marrow megakaryocytes and by circulating platelets before their removal from the circulation.
This understanding provided some rationale for clinical trials of TPO-mimetic agents in patients with ITP. Impetus was provided by the report of a patient with a cyclic variant of ITP who has responded to PEG-rHuMGDF for more than ten years,64,65 and by a Japanese study finding responses to the TPO analog in three of four patients with refractory chronic ITP. 66 (see Table 1).
Rationale for romiplostim use in chronic ITP.

Romiplostim
Romiplostim (Nplate), formerly known as AMG-531, was the first of the second generation TPO-mimetics to enter clinical trials for patients with ITP. It is an Fc-peptide fusion protein (peptibody) that binds to the same site of the TPO receptor binding domain as the native hormone with no sequence homology. 67 Thus, this molecule activates c-mpl without a reasonable worry of inducing antibodies that might cross-react with endogenous TPO. Romiplostim consists of 4 mpl binding peptides linked to an immunoglobulin Fc carrier domain, which binds to the FcRn salvage receptor leading to endothelial recirculation, thus prolonging the half-life of the drug. Romiplostim is produced by recombinant DNA technology in E. Coli. 68
Preclinical development
In a series of in vitro studies by Broudy et al romiplostim stimulated growth and maturation of megakaryocyte colonies from murine marrow cells in a dose-dependent manner. 69 The minimum concentration of romiplostim needed to stimulate megakaryocytic growth was 30 ng/ml, and the maximum effect occurred at a drug concentrations of 1000–2000 ng/ml. Romiplostim in combination with TPO resulted in a greater than additive effect on megakaryocyte colony growth. It also showed synergy with other growth factors such as erythropoietin, IL-3, IL-6 and stem cell factor. 69 Serum-free liquid cultures with romiplostim supported the maturation of megakaryocytes as shown by the development of polyploid megakaryocytes with a predominant DNA content of 32 N and 64 N, identical to that of parallel TPO-stimulated cultures. To determine if romiplostim would activate human mpl receptor, BaF3 cells expressing human mpl receptor were cultured in the presence of TPO and romiplostim. 69 Romiplostim stimulated proliferation of BaF3-hMpl cells in a dose-dependent fashion, and had no effect of BaF3 cells that did not express the mpl receptor. Romiplostim was shown to compete at least as effectively as TPO for the same binding site on mpl. It effectively blocked binding of [125I] TPO to normal human platelets, with 50% inhibition at a romiplostim concentration of 3 to 10 ng/ml. Stimulation of BaF3 cells with romiplostim resulted in tyrosine phosphorilation of the mpl receptor, JAK2 and STAT5, similar to TPO. 69
Romiplostim was effective in stimulating megakaryocyte colony growth in human, baboon and cynomolgus monkey myeloid progenitor cell cultures. 70 In exploratory studies in mice, rats and rhesus monkeys, the drug increased platelet counts and caused no apparent thrombocytopenia after single or repeated intravenous doses.
In live animal studies, romiplostim was able to increase platelet counts in models of chemotherapy- and radiation-induced thrombocytopenia. 70 Mice administered carboplatin chemotherapy and sublethal irradiation were given a single dose of placebo or romiplostim (100 mg/kg). The platelet count dropped to 10% of the baseline level by day 10 in the placebo group, remained less than 100 x 10 9 /L from days 9 through 12, and returned to normal by day 21. Mice treated with romiplostim had nadir counts more than 700 x 10 9 /L occurring between days 5 and 6. Thus, romiplostim significantly reduced the severity and duration of chemo/radiation-induced thrombocytopenia.
Pharmacokinetics
After intravenous administration to 12 healthy volunteers, romiplostim demonstrated non-linear pharmacokinetics for the dose range 0.3 to 10 μg/kg. 67 After an IV dose of 0.3, 1 and 10 μg/kg, the half lives were 1.50, 2.41 and 13.8 hours. The area under the curve increased in a dose dependent manner from 0.964 to 1530 pg.h/ml between doses of 0.3 and 10 μg/kg. The maximum serum concentration also depended on the dose, with concentrations of 2,810 pg/ml and 210,000 pg/ml for doses of 0.3 and 10 μg/kg respectively. The serum level of romiplostim following a subcutaneous dose peaked between 24–36 hours.
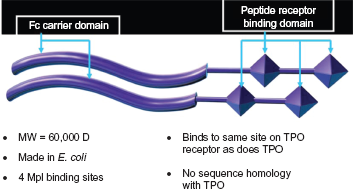
Romiplostim: a Peptibody. Courtesy of D. Kuter.
Clinical development
In the first human, randomized, double-blind, placebo-controlled study, romiplostim was given to 48 healthy adults in single doses from 0.3 to 10.0 μg/kg subcutaneously (SC) and 0.1 to 2.0 μg/kg intravenously (IV). 67 Platelet counts measurably increased 1–3 days after intravenous administration and 4–9 days after SC administration, with peak platelet numbers after 12–16 days. Counts returned to baseline by 28 days. There was a dose-dependent response with a six-fold increase at the maximum dose of 10 μg/kg. In 6 of 8 patients given 2.0 μg/kg subcutaneously, the platelet count doubled. Platelet counts rose to a similar extent after the same dose irrespective of the route of administration.
Thirty healthy subjects in Japan participated in a similar phase I study. 68 They were randomized to receive a single subcutaneous dose of romiplostim (0.3, 1.0 or 2.0 μg/kg) or placebo. In 4 of the 8 subjects receiving 1 μg/kg and 7 of 8 subjects receiving 2 μg/kg, the platelet count increased more than 1.5 fold over baseline.
In a phase I/II, open-label, dose-escalation, multicenter European study, 16 patients with ITP were assigned to one of four sequential dose cohorts of romiplostim: 30, 100, 300 and 500 μg. 71 The drug was given as 2 subcutaneous injections 2 to 3 weeks apart, and patients were followed up for 8 weeks post treatment. The primary and secondary objectives of the study were to evaluate safety and efficacy of romiplostim. Two patients were withdrawn from the study because of unacceptably high platelet counts after the first doses of 300 μg and 500 μg, and the 500 μg cohort was discontinued. Overall, 12 of 15 patients (80%) had an increase in their platelet counts to ≥20 x 10 9 /L above baseline and 8 of 15 patients (53%) increased their platelet counts to ≥100 x 10 9 /L. Achievement of a platelet response was further evaluated after conversion of the unit dose to a weight-based dose. Eight of 11 patients (73%) with a dose equivalent to >1 ug/kg achieved a therapeutic target response defined as a platelet count doubling from baseline and between 50–450 x 10 9 /L.
Comparable results were obtained in a similar, phase I/II open label, dose-finding, multicenter American study in patients with ITP. 11 In the phase I of the study, 24 patients were assigned romiplostim as two subcutaneous doses of 0.2, 0.5, 1.0, 3.0, 6.0 and 10 μg/kg separated by 2–3 weeks. Overall, 7 of 12 patients given 3.0, 6.0 or 10 ug/kg of romiplostim achieved a platelet count above 50 x 10 9 /L, 4 within the target range (50–450 x 10 9 /L) and 3 with platelet counts exceeding 450 x 10 9 /L. In the phase II of this study, 21 patients were randomized to receive weekly subcutaneous injections of romiplostim for 6 weeks (at doses of 1.0, 3.0 or 6.0 μg/kg) versus placebo. 11 In one patient receiving 6.0 ug/kg, the platelet count increased to above 500 x 10 9 /L and the 6.0 ug/kg cohort was closed, leaving a total of 16 patients in the romiplostim arm (8 each in the 1.0 and 3.0 ug/kg) and 4 in the placebo arm. Ten of 16 patients (63%) treated with romiplostim at doses of 1.0 or 3.0 μg/kg achieved the target platelet range.
In Japan, a similar phase II, multicenter, open-label, sequential-cohort, dose-escalation study was designed to evaluate the safety and efficacy of romiplostim in patients with ITP. 72 Four patients were to be enrolled at each of 4 sequential dose levels of romiplostim: 1.0, 3.0, 6.0 and 10.0 ug/kg. When one patient in the 6.0 ug/kg cohort had a platelet count of 980 x 10 9 /L, it was decided not to begin the 10.0 ug/kg cohort. A subcutaneous dose was administered weekly for a total of 2 weeks. Patients who achieved a platelet response (doubling of the baseline platelet counts to a level ≥50 x 10 9 /L) could continue into a treatment-continuation phase, where they received romiplostim weekly with the option to adjust the dose to achieve a platelet count between 50–200 x 10 9 /L. When the treatment-continuation phase ended, the patients were followed up for 3 more weeks after the last dose. Overall, 7 of the 12 patients (58.3%) achieved a platelet response, 4 in the 6.0 ug/kg, 2 in the 3.0 ug/kg and 1 in the 1 ug/kg cohort. Five patients entered the treatment-continuation phase, with platelet counts ≥50 x 10 9 /L maintained in approximately half of the weekly assessments.
These studies were the bases for the registration trials: two parallel, phase III, randomized, placebo-controlled studies comparing romiplostim and placebo in patients with ITP at multiple centers in the United States and Europe. 12 The trials were identical except that one recruited splenectomy failures while the other included only non-splenectomized patients. A total of 63 splenectomized and 62 non-splenectomized patients were randomized (2:1) to romiplostim versus placebo. All had failed at least one prior mode of therapy, and in fact two-thirds of patients had failed three or more prior therapies. The platelet count had to be consistently below 30 x 10 9 /L, and the median platelet count in the studied patients was 14–19 x 10 9 /L. The drug was administered weekly over a 24-week period beginning at 1.0 μg/kg, then titrated to maintain platelet counts 50–200 x 10 9 /L. Initially, the maximum dose allowed was 15 ug/kg. The primary objectives were the efficacy of romiplostim as measured by a durable platelet response (defined as a platelet count more than 50 x 10 9 /L for at least 6 of the last 8 weeks of treatment) and the safety of treatment. A durable platelet response was achieved by 16 of 42 (38%) splenectomized patients give romiplostim versus none of 21 in the placebo group, and in 25 of 41 (61%) non-splenectomized patients given the drug versus 1 of 21 (5%) given placebo. The overall platelet response rate (durable or transient) was achieved in 69 of 83 patients (83%) in patients taking romiplostim compared to 3 of 42 (7%) taking placebo. Reduction or discontinuation of concurrent steroids, danazol or azathioprine by prespecified criteria was achieved in 87% of the romiplostim-treated ITP patients and 38% of those on placebo. Toxicities were tolerable (see below).
A single-arm, open-label extension study is currently evaluating long-term safety and efficacy in patients with chronic ITP treated with romiplostim. 73 Eligible patients had completed a prior phase III romiplostim study. 12 Romiplostim was administered every week subcutaneously, with dose adjustments to maintain platelet counts in the target range (50–200 x 10 9 /L). As of May 2009, 291 adult patients have been treated with romiplostim; thirty percent had undergone prior splenectomy. The median weekly dose across the overall study population was 4 μg/kg. More than half of the patients were able to maintain a sustained platelet count above 50 x 10 9 /L. In the extension trial, 9.2% experienced serious adverse events, but less than 5% of patients discontinued therapy because of these (2 for marrow reticulin fibrosis, one for hemorrhage, one for deep vein thrombosis, and one for the finding of monoclonal gammopathy of uncertain significance).
Health-related quality of life was also evaluated in these patients. It revealed a significant improvement for romiplostim-treated patients in terms of symptoms, activity levels and fear of the disease. 74
Dosage and administration
The recommended starting dose of romiplostim is 1 μg/kg based on actual body weight and given once weekly as a subcutaneous injection. The dose may be adjusted weekly by increments of 1 μg/kg to achieve and maintain a platelet count above 50 x 10 9 /L. The maximum weekly dose should not exceed 10 μg/kg. The drug should be held when platelet count is more than 400 x 10 9 /L. Romiplostim should be discontinued if the platelet count does not increase after 4 weeks at the maximum dose. A complete blood count (CBC) including platelet count should be monitored weekly for at least 2 weeks after discontinuation of romiplostim. 75
Drug interactions
Romiplostim may be used with other ITP therapies, such as corticosteroids, danazol, azathioprine, intravenous immunoglobulin (IVIG), and anti-D immunoglobulin. If the patient's platelet count is greater than or equal to 50 x 10 9 /L, concomitant therapies may be reduced or discontinued. 75 There are no known drug interactions.
Safety/side-effects
Most side-effects reported with romiplostim are mild to moderate. Headache is the most frequent adverse effect reported (35% in the phase III trials versus 32% of placebo patients) and is usually not severe. Fatigue, sore throat and oral mucosal blistering have also been reported.11,12,68,71,72
A few potentially serious concerns deserve special mention. One is “rebound thrombocytopenia” after stopping the drug, seen in a few cases. This may be due to a therapy-induced transient expansion of the platelet and megakaryocyte pool, leading to increased clearance of endogenous TPO. The temporary TPO deficiency state can result in a transient drop in platelet counts below the pre-treatment baseline, with concomitant increased bleeding risks.11,71 Severe bleeding complications have not been common in the trials, but clinicians should be aware of this potential problem. One should monitor closely and be prepared to intervene with rescue therapies; one might consider tapering rather than abruptly discontinuing the drug in responding patients.
Although thromboembolic arterial and venous events have been seen in patients with ITP taking romiplostim, generally there have been other identifiable risk factors for thrombosis, the events have not appeared to be more common than expected, and a majority of the cases occurred with platelet counts less than 400 x 10 9 /L. 12 In the open-label extension study, after 5 years of follow up, thrombotic events were experienced in 17 of 291 patients (6%) and did not seem to increase in frequency over time. 73
Because stimulated megakaryocytes release cytokines such as transforming growth factor B which can stimulate fibroblasts to lay down reticulin, marrow fibrosis has been a potential concern with the use of TPO-mimetic agents. Increased deposition of reticulin fibers has been documented in some patients receiving romiplostim. It is distinct from collagen fibrosis (trichrome staining) which is less reversible and usually associated with myeloproliferative disorders or tumors metastasic to the bone marrow. In rats, romiplostim produced a dose-dependent increase in bone marrow fibrosis that resolved after treatment withdrawal. 76 The doses of romiplostim required were up to 10 times the recommended dose for treatment of ITP patients. Kuter reported on reticulin deposition from a retrospective analysis of 271 romiplostim-treated chronic ITP patients and from a small prospective study in which bone marrow biopsies were obtained before and after romiplostim initiation. 76 In the retrospective study, 11 of 271 treated patients had a bone marrow biopsy, and 10 of the 11 had some reticulin deposition; 6 of these were receiving doses of romiplostim higher than currently recommended. The prospective study involved 10 patients and only one had a clear increase in reticulinformation with treatment. The authors conclude that romiplostim produces reversible, dose-dependent bone marrow changes in animals as well as relatively benign increases in bone marrow reticulin in some patients with ITP, which are reversible when treatment is discontinued. These changes are distinct from a true myeloproliferative disorder, and generally there has been no effect on blood counts. In the 5 year follow up extension study, 9 of 291 patients had reticulin present or increased with no evidence of progression to collagen fibrosis or chronic idiopathic myelofibrosis. 73
Neutralizing antibodies against romiplostim have been rarely reported.73,77 No associated thrombocytopenia from the antibodies have been observed because the antibodies do not react against endogenous TPO.
Use in special populations
There is no experience with romiplostim use in pregnant women. In animal reproduction and developmental toxicity studies, romiplostim crossed the placenta, and adverse fetal effects included thrombocytosis, post-implantation loss, and an increase in pup mortality. Therefore, romiplostim should be used during pregnancy with great caution, only when the potential benefit to the mother justifies the potential risk to the fetus. 75 It is not known whether romiplostim is excreted in human milk, but human IgG is known to be excreted in milk in at least small amounts. Because of the potential for serious adverse reactions in nursing infants from romiplostim, a decision should be made to either discontinue nursing or discontinue the drug. 75
Of the 271 patients who received romiplostim in ITP clinical studies, 55 (20%) were age 65 and older, and 27 (10%) were age 75 and older. No overall differences in safety or efficacy were observed between elderly or younger patients in the placebo-controlled studies. 75
No clinical studies have been conducted in patients with renal or hepatic impairment. Therefore, romiplostim should be used with caution in these populations. 75
Regulatory agency approval
Based on the efficacy and safety data, romiplostim was approved on August 22, 2008 by the US Food and Drug Administration (FDA) for use in ITP patients who have failed at least one prior therapy, including corticosteroids, high dose intravenous immunoglobulin, or splenectomy. 78 Soon after, the drug was also approved by regulatory agencies in the European Union and Australia.
Approval by the FDA was accompanied by substantial pharmacovigilance efforts including a Risk Evaluation and Mitigation Strategy (REMS), a management plan that utilizes tools beyond approved package insert to mitigate serious risks and ensure that the benefits of the drug outweighs the risks. The main two risks included in this risk management plan are bone marrow reticulinformation and severe thrombocytopenia following discontinuation of the drug. Possible risks without signals from clinical study data included thrombosis and progression of disease in patients with myelodysplastic syndromes (MDS). No risk of hematologic malignancies was observed in the phase III studies.
Eltrombopag and other TPO-Mimetics
Eltrombopag (Promacta; GlaxoSmithKline) is the other second generation TPO-mimetic which has completed phase II/III studies. It is a synthetic non-peptide orally-available small molecule that activates JAK2 and STAT 5 pathways via binding to the trans-membrane portion of c-mpl, unlike native TPO and romiplostim. 79 This difference in binding sites raises at least theoretic possibilities that some subjects might respond to one but not the other TPO-mimetic, or that the two drugs could show enhanced effect when used together.
Eltrombopag was evaluated in a phase II and phase III randomized, double-blind, placebo-controlled, multi-center trials for ITP patients that had failed at least one prior therapy, trials with very similar design to the romiplostim studies. 79 One-hundred and eighteen adult patients with chronic ITP were randomized to receive 30, 50 or 75 mg doses of eltrombopag or placebo. The primary endpoint (a platelet count ≥50 x 10 9 /L on day 43) was achieved in 28%, 70% and 81% of patients receiving 30, 50 and 75 mg of eltrombopag, compared to 11% of patients on placebo.
In the phase III RAISE trial, a multicenter study involving adults from 63 sites in 23 countries, 114 adult ITP patients were randomized to receive eltrombopag (50 mg) versus placebo. 80 A platelet response, defined as platelet count ≥50 x 10 9 /L on day 43 of treatment, was seen in 59% of patients with eltrombopag compared to 16% with placebo. Thirty-six percent more of the patients had “clinically meaningful platelet responses,” and like romiplostim, response was not predicted by baseline platelet count or splenectomy status. The drug was generally well-tolerated with an adverse events profile which generally did not differ significantly from that with placebo. In this phase III trial, liver function abnormalities were encountered in 10% of eltrombopag-treated patients versus 8% on placebo, resulting in the requirement for a black box warning on the label.
Eltrombopag was approved by the FDA on November 20, 2008 for adults with chronic ITP who have had insufficient response to corticosteroids, immunoglobulins or splenectomy. 81 The recommended starting dosage of eltrombopag is 50 mg/day, with a maximum dosage of 75 mg/day. 82 Like romiplostim, it was approved with a REMS, where the most serious safety concerns identified in clinical trials included hepatic toxicity as well as worsening thrombocytopenia upon the drug's discontinuation. Signals of potentially serious risks include bone marrow reticulinformation leading to fibrosis, a risk for hematologic malignancy and progression of disease in patients with MDS, and a risk for thrombosis. 81
A number of other TPO-mimetics are in development. 83 The furthest along of these is AKR-501, an oral agent currently in human ITP trials. 84
Current and Future Roles for Romiplostim
For the few ITP patients with severe and highly refractory disease, romiplostim can be miracle drug. It is less clear whether romiplostim and other TPO-mimetics will quickly move up the therapeutic algorithm for earlier use in ITP, although this is the opinion of many experts. One needs to consider that with previously available therapies, about 98% of ITP patients have had a favorable outcome in terms of maintaining safe platelet counts and avoiding serious bleeding, but not infrequently this is achieved at high costs of side-effects, risks, psychological trauma, and sometimes monetary costs. TPO-mimetic therapy is attractive for the high response rates, favorable tolerability profile and lack of immunosuppression. One major drawback is the need for ongoing therapy, in contrast to such modalities as rituximab or splenectomy where durable unmaintained remissions are more common. Many ITP treatments are costly, and in the case of romiplostim the current wholesale acquisition price is $1,083 for a single-use vial of 250 μg which would equate with the weekly cost for about half of treated patients.
In choosing between TPO-mimetic agents in ITP, there have been no head-to-head trials of romiplostim and eltrombopag, making firm conclusions difficult about relative efficacy and safety except to say that reported response rates were similar in the similarly designed trials. The most obvious difference between the two agents is the route of administration, and while most patients might prefer the convenience of an oral agent, this introduces a number of issues of possible absorption issues, patient compliance issues, self- administration versus need to travel to a medical facility, and cost/reimbursement differences. Bone marrow fibrosis has not emerged as a serious problem with either agent, but it is difficult to know whether this might be more common with one agent than the other because the incidence of subclinical changes largely reflects how hard one looks for them. Similarly, while mild to moderate degrees of uncomfortable symptoms such as dizziness, myalgia, and arthralgia were recorded more often in romiplostim studies, it is hard to be sure whether these really differ between the two agents without concurrent assessment. A risk of thromboembolic events may accompany any successful treatment of ITP, and an obvious difference between agents has not been appreciated. It might be prudent at present to prefer romiplostim in a hepatically-impaired patient, but the true incidence and severity of hepatic risks remain to be clarified. Also uncertain are the advantages and disadvantages of newer TPO-mimetics as they proceed in development.
Romiplostim and other TPO-mimetic agents are under study in a wide range of other thrombocytopenic disorders. Although the sum of trials of first generation TPO-mimetics for chemotherapy-induced thrombocytopenia can be characterized as disappointing, this remains an area of active study with hopes that unmet needs can be identified and addressed. Phase III trials are underway with romiplostim both as treatment for those patients with myelodysplastic syndromes whose thrombocytopenia is a major issue, and also as an adjunct to support others with myelodysplasia through myelotoxic therapies. Stem cell transplant uses are being explored including mobilization and post-transplant thrombocytopenia. With advanced liver disease, thrombocytopenia is usually moderate, but can frequently limit effective anti-viral therapy for hepatitis C, and there may also be a role for TPO-mimetic agents in some liver transplant settings. Routine platelet donations would probably not utilize expensive drugs which require one to two weeks for effect, but TPO-mimetics might prove useful for special platelet donors (rare antigens, matched repeated donors).
Conclusion
Romiplostim is a potent TPO-mimetic agent, binding to mpl to predictably raise platelet counts in normal volunteers and those with a number of thrombocytopenic disorders. In chronic ITP, it has become clear that blunted platelet production is an important component of the pathophysiology, and romiplostim is highly effective at correcting this. It is attractive for earlier use in the majority of chronic ITP patients who cannot be maintained at a satisfactory level with corticosteroids, IVIG or anti-RhD. Potential concerns have included increased reticulinformation, risks for thromboembolic complications, and risks of “rebound thrombocytopenia,” but these have not emerged as a major problem. Real drawbacks to increasing use include the need for indefinite therapy, costs, and an increasing number of effective alternatives. The ability of this drug and its analogs to fulfill needs in other thrombocytopenic disorders remains under very active clinical study.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. Dr. Rice has received honoraria for speaking on romiplostim from Amgen and for speaking on eltrombopag from GSK. The peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.