
Abstract
Coronary artery disease (CAD) is a major cause of morbidity and mortality in the world. Therapy for stable CAD is currently based on conventional medical therapy, including nitrates, β-blockers and calcium-channels antagonists and, more recently, metabolic therapy, of which a pivotal therapeutic role is increasingly recognized. Under normoxic condition, the healthy heart derives 2/3 of its energy from the free fatty acid (FFA) pathway, the other source of energy being derived from glucose oxidation. However, glycolysis requires less O2 per mole of ATP generated compared with FFA oxidation. On this basis, shifting energy substrate utilization from fatty acid metabolism to glucose metabolism can be more efficient in terms of ATP production per mole of oxygen utilized. A number of different approaches have been used to manipulate energy metabolism in the heart. These approaches include direct agents, such as dichloroacetate, L-carnitine, ribose or lipoic acid which directly increase glucose oxidation, or indirect methods, through the inhibition of free fatty acids oxidation. Among these, the most important are carnitil-palmitoyl-transpherase I (CPT-I) inhibitors, which inhibit FFA mitochondrial uptake (e.g. etomoxir, perhexiline, oxphenicine), or 3-ketoacyl-coenzyme-A thiolase (3-KAT) inhibitors, such as trimetazidine, which inhibits the last enzyme involved in β-oxidation. In most patients with ischemic heart disease metabolic abnormalities, if not adequately treated, will heavily contribute to the occurrence of complications, of whom severe left ventricular dysfunction is at present one of the most frequent and insidious. In this paper, all possible metabolic approaches to ischemic heart disease are reviewed and discussed.
Introduction
Ischemic heart disease is a major cause of morbidity and mortality in the world. Although revascularization has proven to reduce myocardial infarction (MI) and death in patients with acute coronary syndromes (ACS),1,2 the same is not true for chronic stable angina. 3 Therapy for stable coronary artery disease (CAD) is currently based predominantly on medical therapy, with revascularization serving an adjunctive role, 4 and has two major focuses. The first is a vasculoprotective role, aimed at delaying atherosclerotic disease progression, thereby reducing future cardiovascular events and death and improving quantity of life. The second is an anti-ischemic (antianginal) role, focused on improving the ischemic imbalance, thereby reducing the severity and frequency of anginal symptoms and, again, improving quality of life.
However, myocardial ischemia is the metabolic consequence of an inadequate blood supply to the myocardium. It is basically a metabolic problem, leading to an imbalance of normal metabolic pathways through which normal heart is able to produce energy. Under normoxic condition, the healthy heart derives most of its energy from the free fatty acid (FFA) pathway that accounts for approximately two thirds of energy production (adenosine-triphosphate, ATP), the other source of energy being derived from glucose oxidation and lactate.5–7 In hypoxic conditions, myocardial cells respond to mild to moderate ischaemia by accelerating glucose uptake to generate sufficient ATP in order to maintain ionic gradients and calcium homeostasis, as glycolysis requires less O2 per mole of ATP generated compared to FFA oxidation. On the other hand, severe ischaemia rapidly induces an imbalance between the requirement of cardiac tissue for oxygen and coronary blood supply, resulting in functional, metabolic, and morphological alteration of the myocardium, including arrhythmias, contractile failure and electrophysiological abnormalities. At a cellular level, glucose uptake is decreased and conversion to lactate is increased; lactate uptake by the heart is switched to lactate production, and pyruvate is mostly transformed into lactate, thereby increasing cell acidosis. The free fatty acid pathway is slowed down, resulting in less ATP production. These metabolic changes lead to disruption of cell homeostasis, alterations in membrane structure, and ultimately cell death.
Given this pathophysiological background and the difficulty of classic haemodynamic agents to control the total ischemic burden in some patients, it seems logical to consider pharmacological manipulation of cardiac energy metabolism as an adjunctive therapeutic option. Optimization of cardiac energy metabolism is based on promoting cardiac glucose oxidation. This has been proven to enhance cardiac function and protect myocardial tissue against ischaemia-reperfusion injury. 8
Stimulation of myocardial glucose oxidation can be achieved either directly with stimulation of glucose metabolism, or indirectly through inhibition of fatty acid β-oxidation, in order to shift energy substrate utilization away from fatty acid metabolism and towards glucose metabolism, which is more efficient in terms of ATP production per mole of oxygen utilized. In fact, increasing utilization of glucose and lactate, which are more efficient fuels for aerobic respiration, the oxygen consumption efficiency of the myocardium can be improved from 16 to 26%. 8 Additionally, heart and arm skeletal muscle glucose uptakes are inversely related to serum FFA levels 9 and increased FFA flux from adipose tissue to non-adipose tissue amplifies metabolic derangements that are characteristic of the insulin resistance syndrome, 10 common pattern in patients with ischemic heart disease. New findings also suggest that raised FFA levels not only impair glucose uptake in heart and skeletal muscle but also cause alterations in the metabolism of vascular endothelium, leading to premature cardiovascular disease. 11 Therefore, metabolic therapy could also play a beneficial role in terms of glucose metabolism homeostasis.
A number of different approaches have been used to manipulate energy metabolism in the heart; these include:
indirect agents, trough the inhibition of free fatty acids oxidation; among these, the most important are carnitine-palmitoyl-transpherase I (CPT-I) inhibitors, which inhibits FFA mitochondrial captation (e.g. etomoxir, perhexiline, oxphenicine), or 3-ketoacyl coenzyme-A thiolase (3-KAT) inhibitors, such as trimetazidine, which inhibits the last enzyme of FFA β-oxidation.
direct agents, such as dichloroacetate, ribose and lipoic acid which stimulate directly glicolysis, or L-carnitine which increases glucose oxidation despite elevated FFA levels.
CPT-I inhibitors (etomoxir, perhexiline, oxfenicine)
Etomoxir, perhexiline and oxfenicine are CPT-I inhibitors, the key enzyme for mitochondrial FFA uptake; its inhibition, therefore, reduces FFA oxidation and their inhibitory effect on pyruvate dehydrogenase. As a consequence, glucose oxidation is increased12,13 (Fig. 1). Etomoxir, initially developed as an antidiabetic agent, has then been observed to improve left ventricular performance of pressure-overloaded rat heart. 14
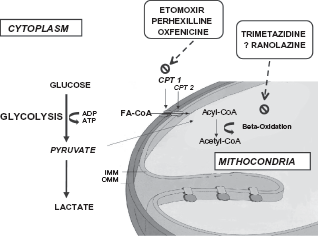
Mechanisms of action of metabolic agents on myocardial metabolism. Perhexilline, oxfenicine, and etomoxir prevent uptake of free fatty acids by inhibiting carnitine palmitoyltransferase (CPT) I, which is a key mitochondrial enzyme involved in this process. Trimetazidine and, possibly, ranolazine, inhibit beta-oxidation of free fatty acids. These actions shift myocardial substrate use from free fatty acids to glucose, which is more efficient in terms of energy production, leading to an oxigen-sparing effect.
These effects have been considered due to a selective modification of gene expression of hypertrophic cardiomyocytes. 15 Etomoxir could also increase phosphatase activation, have a direct effect on peroxisome proliferator activated receptor-alpha and up-regulate the expression of various enzymes involved in beta-oxidation. 15 The first clinical trial employing etomoxir has shown a significant clinical and cardiac function improvement in heart failure patients. 16 In experimental animal studies, etomoxir has also been shown to improve glucose metabolism. 17 Additional data have shown that the beneficial effect of etomoxir on reperfusion recovery of ischemic hearts is not due to a lowering of long chain acylcarnitine levels. Etomoxir may improve recovery of function by overcoming fatty acid inhibition of glucose oxidation. 18 However, the use of etomoxir may be limited by the the observation that it may cause cardiac hypertrophy 19 and oxidative stress. 20
Analogously to etomoxir, oxfenicine and perhexiline, originally classified as calcium antagonists, reduce cardiac utilization of long chain fatty acids by inhibiting CPT-I.21–23 They have been initially developed as antianginal agents.24,25 The anti-anginal efficacy of perhexiline was demonstrated in older trials, even in patients not controlled with other antianginal drugs.26,27 Moreover, the drug has also been employed in patients with ischemic heart failure. In a recent study in patients with chronic heart failure, metabolic modulation with perhexiline improved O2 max, left ventricular ejection fraction, symptoms, resting and peak stress myocardial function, and skeletal muscle energetics. 28
More recently, perhexiline has been shown to provide symptomatic relief in the majority of patients with heart failure and angina, with minimal side effects or toxicity. 29 Therefore, CPT-I inhibitors may represent a novel treatment in stable CAD patients. They show a good safety profile, provided that the dosage is adjusted according to plasma levels. Infact, both etomoxir and perhexiline should be used with caution because of reports of cardiac hypertrophy, 30 oxidative stress, 30 hepatotoxicity 30 and peripheral neuropathy30,31 if overdosed.
3-KAT inhibitors (trimetazidine)
A new class of metabolic agents, known as the 3-ketoacyl coenzyme A thiolase (3-KAT) inhibitors, is able to elicit an increase in glucose and lactate combustion secondary to partial inhibition of fatty acid oxidation, producing consistent clinical benefits in patients with ischaemic heart disease.
The most studied agent of this class of metabolic drugs is trimetazidine. Experimental evidence indicates that its mechanism of action is predominantly based on partial inhibition of mitochondrial long-chain 3-ketoacyl coenzyme A thiolase, the last enzyme involved in β-oxidation; 32 however, this issue is still under debate.33,34
Reduction of FFA oxidation determines an increase of glucose oxidation, restoring coupling between glycolysis and carbohydrate oxidation, which leads to ATP production with less oxygen consumption. 35 Furthermore, by stimulating membrane phospholipid turnover during ischemia and reperfusion, trimetazidine redirects FFA toward phospholipids, increasing cell tolerance to ischemia-reperfusion damage.35–37 The anti-ischemic properties of trimetazidine are independent from hemodynamic changes and are associated with a greater recovery of mechanical function after ischemia. 35
Whatever its mechanism of action, the efficacy of trimetazidine in the treatment of angina pectoris has been evaluated in a number of studies, as monotherapy or in combination, in acute and chronic administration, as initial treatment or in patients resistant to β-blockers or Ca2+ channel antagonist.38–60
The effects of acute administration of trimetazidine were initially tested in patients with chronic stable effort angina during exercise testing. Trimetazidine increased effort tolerance and delayed the appearance of ischaemic symptoms and ECG changes. 40 Chronic treatment with trimetazidine confirmed the benefits observed after acute administration and was well-tolerated by the patients. In fact, no appreciable side-effects were recorded, including no significant changes of heart rate and/ or aortic pressure.41–43 Comparison studies have shown that the improvement in ischaemic threshold and exercise tolerance with trimetazidine is similar to that achievable with propanolol and nifedipine, with the additional bonus of a lower incidence of side-effects.44,45
Direct evidence that the effects of trimetazidine are additive to the effects of ‘haemodynamic’ agents was given by a multicentre, randomized, double-blind study where the addition of trimetazidine to propranolol was compared with the addition of nitrates to propranolol. This study was performed in patients with chronic effort angina and documented coronary artery disease and demonstrated that trimetazidine–propranolol was more effective and better tolerated than nitrates combined with propranolol. 46 As a matter of fact, this study showed that only the addition of trimetazidine to the β-blocker was followed by an improvement; conversely the addition of nitrates to the β-blocker had no effect on symptoms and exercise capacity. Along this line of evidence, it is worth mentioning another study where the effects of trimetazidine were assessed in angina patients uncontrolled by diltiazem. 47 This double-blind, randomized study also showed that the addition of trimetazidine to full dose diltiazem resulted in a significant reduction in the number of ischaemic attacks, prolonged the time to 1 mm S–T depression, increased the time to angina during exercise, and increased the maximum work at peak exercise. These beneficial effects were obtained without yielding adverse haemodynamic events or increased side-effects. In another study, 12-week treatment with trimetazidine and metoprolol again significantly reduced clinical symptoms compared with placebo and metoprolol in patients with stable effort angina whose angina was not controlled by metoprolol alone. 48
The Trimetazidine in Poland (TRIMPOL-I) study showed that 4 weeks of treatment with trimetazidine significantly decreased the number of anginal episodes and improved myocardial ischemia and exercise capacity in patients with diabetes. 49
Marazzi et al have shown that in patients with diabetes and chronic stable angina the addition of trimetazidine to standard medical therapy reduces the number of episodes of ST-segment depression, the episodes of silent ischemia, and total ischemic burden. 50
Our group has also confirmed that trimetazidine is effective in differet settings of stable coronary disease53,54 and has extended its use in patients with both, ischemic and non ischemic, heart failure.55,56 Indeed, in these contexts, trimetazidine has been shown to improve symptoms, left ventricular function and, consequentely, quality of life. The main mechanism of action is likely based on trimetazidine-induced increased myocardial cellular reserve. 57 However, additional mechanisms may be represented by improved endothelial function, 58 increased insulin sensitivity 59 and, possibly, intrinsic electrophysiological effects. 60 Nevertheless, no clear-cut impact on mortality has ever been shown with trimetazidine in any specific cardiac condition. Therefore, prospective, randomized, controlled studies are warranted in order to ascertain the exact role of trimetazidine in cardiovascular pharmacology. At present, according to the European Society of Cardiology, its use is advised as an adjunctive treatment in patients with angina not completely controlled by standard hemodynamic therapy. 61
Late inward sodium channel blockers (ranolazine)
Ranolazine, a more recent antiischemic drug, is an active piperazine derivative. The molecular mechanism of action is probably related to inhibition of the late inward sodium channel (late INa),62,63 which pathologically remains open in a wide variety of adverse stimuli to the myocardium (myocardial ischemia, hypertrophy, oxidative stress). This intracellular overload of sodium ions leads to several major contractile, metabolic and electrophysiologic disturbances. 63 Ranolazine appears to play an important role in late INa channels in a concentration-, voltage-, and frequency-dependent manner.62–64 By comparing studies on patients affected by different heart disease, it appeared that the effect of ranolazine on late INa is more pronounced in ischemic or failing myocytes in which the current is amplified. 65 Ranolazine in the immediate-release formulation significantly reduced anginal episodes and nitroglycerin use, and significantly improved exercise duration and time to exercise-induced myocardial ischemia 66 in patients with stable coronary disease. The Monotherapy Assessment of Ranolazine in Stable Angina (MARISA) trial investigated the effect of the sustained-release formulation of ranolazine versus placebo showing a significant, dose-dependent increase in exercise duration, exercise time to angina, and exercise time to 1-mm ST segment depression at trough and at peak drug effect compared with placebo, in patients affected by reproducible treadmill-induced myocardial ischemia and angina. 67 The Combination Assessment of Ranolazine in Stable Angina (CARISA) trial was designed to evaluate the effect of the addition of ranolazine to antianginal drugs, including atenolol, diltiazem, or amlodipine. 68 Also in this study, ranolazine induced a significant reduction in anginal frequency and nitroglycerin consumption, and an increase in exercise duration, time to angina, and time to 1-mm ST-segment depression compared with placebo both at trough and peak drug effect. Furthermore, the patients in CARISA treated with ranolazine who were also diabetic, experienced a significant reduction in hemoglobin A1c compared to placebo although the mechanism of this improvement is not clear.
A more recent study (Efficacy of Ranolazine in Chronic Angina -ERICA- trial) 69 confirmed that the addition of ranolazine to antianginal therapy is associated with a significant reduction in weekly anginal episodes and nitroglycerin consumption, especially in those patients who were more symptomatic. Additionally, the electrophysiological properties of ranolazine have been recently analyzed, and a direct antiarrhythmic effect has been hypothesized. 61 However, despite its preclinical promise, there is currently limited published clinical trial data to suggest an antiarrhythmic effect for ranolazine. As shown in the Metabolic Efficiency with Ranolazine for Less Ischemia in Non-ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) trial, 70 which addressed patients with acute coronary syndrome, episodes of nonsustained supraventricular tachycardia and brief runs of ventricular tachycardia were reduced in patients within the first 7 days of treatment with ranolazine compared with placebo. However, sustained arrhythmias such as new onset atrial fibrillation and ventricular tachycardia were not significantly altered, and ranolazine did not affect hard end points of cardiovascular death, myocardial infarction, or recurrent ischemia over 1 yr of treatment. What the clinical data do indeed suggest is that ranolazine is safe in patients with a normal QT interval. Nevertheless, further studies are needed to establish the specific antiarrhythmic role(s) of ranolazine. 71
Finally, ranolazine may be associated with gastrointestinal disorders (constipation, nausea, vomiting) and dizziness, 72 and is metabolised by the cytochrome P450 isoenzymes CYP 3A4 and CYP 2D6. 73 There is, therefore, a high risk of pharmacokinetic interactions. There is also a risk of pharmacodynamic interactions with drugs that prolong the QT interval. 74 A recent study has shown that long-term therapy with ranolazine seems well tolerated in high-risk chronic stable angina patients, with evidence that symptomatic improvements attributable to ranolazine are not offset by increased mortality. 75 On this ground, together with the fact that comparative trials have failed to show whether ranolazine has a clear-cut impact on mortality, the efficacy of ranolazine in the prevention of angina attacks needs to be reassessed by further studies. 76
As for trimetazidine, at present, according to the European Society of Cardiology, its use is advised as an adjunctive treatment in patients with angina not completely controlled by standard hemodynamic therapy. 61
Dichloroacetate
Dichloroacetate acts by inhibiting fatty acid oxidation and promoting glucose metabolism during periods of elevated free fatty acid concentration associated with myocardial ischemia. It activates pyruvate dehydrogenase complex, a group of enzymes at the inner mitochondrial membrane, rate-limiting step of glucose oxidation. Pyruvate dehydrogenase is inactivated when phosphorylated by pyruvate dehydrogenase kinase. Dichloroacetate increases the activity of pyruvate dehydrogenase complex by inhibiting pyruvate dehydrogenase kinase. 77
In experimental studies, treatment with dichloroacetate has not been shown to reduce infarct size. 78 However, in an experiment by Wargovich, 79 i.v dichloroacetate improved left ventricular stroke volume and decreased systemic vascular resistance and myocardial efficiency index in human subjects with coronary artery disease. No changes were seen in left ventricular end-diastolic pressure, left ventricular dP/dtmax, coronary sinus flow, coronary resistance, or myocardial oxygen consumption.
In patients with NYHA functional class III–IV congestive heart failure, 30 minutes dichloroacetate administration has been observed to stimulate myocardial lactate consumption and improve left ventricular mechanical efficiency, stroke volume and left ventricular minute work, with a simultaneous reduction in myocardial oxygen consumption. 80 However, an intravenous infusion of the same dose of dichloroacetate over 15 minutes in patients with heart failure and ejection fraction ≤40% has not been shown to be associated with improvement in non-invasively assessed left ventricular function. 81 In preclinical studies, McVeigh et al showed that dichloroacetate stimulation of glucose oxidation improves recovery of ischemic rat hearts. 82 At present, the clinical use of dichloroacetate is limited by its low power and its short half-life, conditions that do not allow its clinical use. 83
L-carnitine
L-carnitine is an essential cofactor of fatty acid metabolism, shuttling the end-products of peroxisomal fatty acid oxidation into the mitochondria and modulating the intramitochondrial acyl-coenzyme A/coenzyme A ratio. Although its main role is enhancement of FFA metabolism, experimental evidence also supports an enhancement of glucose metabolism. Administration of the related propionyl-L-carnitine to the injured rat myocardium results in improved functional recovery and glucose use, supporting the theory that L-carnitine's beneficial effects are due to its ability to increase glucose oxidation despite elevated FFA levels.84,85
Several human and animal studies support a modest benefit in terms of left ventricular energetics and function with L-carnitine administration.84,85 A direct antiischemic effect of this drug has also been hypothesized. A crossover study designed to compare the long-term anti-ischaemic effects of oral L-propionylcarnitine with diltiazem in patients with stable, exercise-induced angina evidenced that both L-propionylcarnitine and (high-dose) diltiazem result in anti-ischaemic effects and decrease anginal attacks in daily life. Nevertheless the effect of diltiazem on exercise-induced ischaemia appears more pronounced than that of L-propionylcarnitine. 86 In a subsequent study L-propionylcarnitine was shown to induce only marginal additional antiischemic effects in anginal patients who were still symptomatic despite maximal conventional antianginal therapy. 87 More recently Iyer et al 88 examined the efficacy of oral carnitine vs placebo in 47 patients with chronic stable angina in improving exercise tolerance. There was no improvement in time to ST depression, ST score, or rate-pressure product. Nevertheless, the carnitine-treated group demonstrated significant improvements in exercise duration and time needed for ST changes to return to baseline.
Najafi et al have recently demonstrated an antiarrhythmic effect of L-carnitine in isolated rat hearts subjected to ischemia: two-hour perfusion with L-carnitine during ischemia/reperfusion markedly and dose-dependently decreased the incidence of ventricular tachycardia, ventricular fibrillation, and ventricular fibrillation duration. 89 Similar datas were collected by other groups. 90 These results confirm previous preliminary observations on the antiarrhythmic effect of L-carnitine in humans with ischemic heart disease. 91
Nevertheless, considering the lack of significant clinical studies, the cardiological community has not yet advocated the use of carnitine in patients with ischemic heart disease.
Mitochondrial metabolic oxidants: lipoic acid and coenzyme Q
Lipoic acid is a cofactor of the pyruvate dehydrogenase enzyme system while coenzyme Q (ubiquinone) is a mitochondrial coenzyme which is essential for the production of ATP. The presence of these molecules indicates that mitochondria can defend themselves against harmful effects of the oxygen atmosphere.92,94 Lipoic acid is synthesized both in the animal and in the human body. 95 This fatty acid with eight carbon atoms containing disulphide groups at the sixth and eighth carbon atoms closed in a pentamerous ring is essential for the function of mitochondrial pyruvate dehydrogenase, thus enhancing glucose metabolism. Both lipoic acid and its reduced form are excellent metabolic antioxidants, as they take part in the antioxidant redox cycle of the organism. 96
Coenzyme Q, also called ubiquinone or ubidecarenone, in its reduced form is an excellent antioxidant. The name “ubiquinone” also indicates that it can occur in various places. The endogenous coenzyme Q can capture perferril, carbon-centred lipids, lipid-peroxyl and alkoxyl radicals. Coenzyme Q has been shown to be beneficial due to its inhibitory effect on lipid peroxidation and on the oxidation of endogenous coenzyme Q-9 as well as by improving mitochondrial respiration.94–97 CoQ10 also decreases blood viscosity and improves blood flow to cardiac muscle in patients with ischemic heart disease. 98 In ischemic heart disease, it has been shown to decrease the spill-over of inflammatory cytokines and prevent hypergycemia induced endothelial cell damage, monocyte adhesion and evolution of atherosclerotic lesions in HUVEK (human umbilical vein endothelial cells) from diabetic patients. 99 In this context, CoQ10 has recently been shown to be effective in enhancing endothelial functionality in patients with coronary disease by counteracting nitric oxide oxidation. 100 Several controlled trials in patients with ischemic heart disease showed significant improvement in exercise tolerance, reduction of ST-segment depression and angina, with no alteration in heart rate or blood pressure.101,102 As for most metabolic agents, also CoQ10 could be particularly effective in patients with heart failure, as recently shown in a double-blind, placebo-controlled cross-over study. 103 Nevertheless, for CoQ10 therapy implementation, further study are warranted, for both coronary disease and heart failure.
Ribose
Ribose is a pentose sugar that has been shown in numerous animal experiments to enhance ATP production and improve cardiac function. Ribose can enhance metabolism by entering the pentose phosphate pathway and bypassing the rate limiting enzymes of glucose-6-phosphate dehydrogenase and 6-posphogluconate-dehydrogenase, thus enhancing glucose metabolism. Furthermore, ribose has no influence on coronary blood flow, myocardial oxygen consumption, and hemodynamics in the normal heart.
Pliml et al 104 studied 20 male patients with documented severe coronary artery disease. After an initial treadmill test, the subjects whose stress test showed reproducibility were randomized to receive four daily doses of ribose vs placebo. On day 5, the treadmill exercise test was repeated. The ribose-treated group showed significant improvement in time to ST segment depression and time to moderate angina. These changes were not significant in the placebo group.
Omran et al 105 studied the benefits of ribose in 15 patients with stable chronic coronary disease and NYHA class II or III CHF. Patients were given ribose or placebo (dextrose) for a 3-week treatment period. After a 1-week washout period, the alternative treatment was administered for another 3 weeks. The investigators reported significant improvements in quality of life assessed with a questionnaire and functional capacity assessed by using an exercise ergometer. However, peak exercise capacity was not affected. Patients were also evaluated echocardiographically before and after the treatment periods. No difference was found in left ventricular volume, stroke volume, or ejection fraction. Ribose treatment, however, resulted in significant deceleration of the E-wave, with significantly smaller left atrial volume and higher left atrial contribution of ventricular filling. The authors hypothesized that in presence of ATP deficiency, calcium may bind to troponin longer during diastole. Treatment with ribose hence improves diastolic relaxation by restoring ATP levels.
Similarly to other metabolic drugs, a more recent study has shown that ribose can also improve the ventilatory exercise status in advanced heart failure patients. 106 In fact, D-ribose may provide the necessary metabolic substrate to benefit the energy-deficient state found in heart failure.
Finally, no adverse events have been reported with ribose and this nutriceutical does not appear to affect hemodynamics, 104 making it a very attractive adjunctive treatment in patients usually on politherapy. However, its exact role in the pharmacological armamentarium needs yet to be defined.
Conclusion
Modulation of myocardial metabolism is a novel, attractive approach to protect cardiac cells and to improve performance of dysfunctioning myocardium in acute and chronic ischemic heart disease, as well as in the setting of chronic heart failure.
However, at the best of our knowledge, there are no double-blind, randomized clinical trials demonstrating a reduction in mortality with conventional plus metabolic therapy versus metabolic therapy alone. It would be useful to assess whether this novel approach could have beneficial effect in terms of mortality, in patients with coronary artery disease.
Given the available evidence, metabolic therapy deserves to be regarded as an exciting alternative/ addition to classic haemodynamic agents and opens new promising therapeutic opportunities.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest.