
Abstract
Tipranavir (TPV) is a selective nonpeptidic HIV-1 protease inhibitor (PI) which is used in the treatment of treatment-experienced adults with HIV-1 infection. Tipranavir is administered orally twice daily in combination with low-dose ritonavir. The durable efficacy of tipranavir, in combination with low-dose ritonavir (tipranavir/ritonavir 500 mg/200 mg twice daily), has been demonstrated in well designed trials in treatment-experienced adults infected with multidrug-resistant strains of HIV-1. In treatment-experienced adults with HIV-1 infection receiving an optimized background regimen, viral suppression was greater and immunological responses were better with regimens containing tipranavir/ritonavir than with comparator ritonavir-boosted PI-containing regimens. The efficacy appeared to be more marked in patients receiving two fully active drugs in the regimen, with the combination of tipranavir/ritonavir and enfuvirtide (for the first time) appearing to be the most successful. Although tipranavir is generally well tolerated, clinical hepatitis and hepatic decompensation, and intracranial hemorrhage have been associated with the drug. Tipranavir also has a complex drug interaction profile. Thus, tipranavir, administered with ritonavir, is an effective treatment option for use in the combination therapy of adults with HIV-1 infection who have been previously treated with other antiretroviral drugs.
Keywords
Introduction
With the introduction of highly active antiretroviral therapy (HAART) a reduction in HIV-1-related morbidity and mortality has been achieved during the last decade. 1 The aim of antiretroviral therapy (ART) is the achievment of complete viral supression (plasma HIV-1 RNA levels < 50 copies/ml) irrespective of whether the patient is treatment-naïve or treatment-experienced, as this outcome is associated with long-term clinical benefit. 2 But virological failure remains a major problem for many patients especially those who received extensive previous antiretroviral treatment or have multidrug-resistance virus. 3 Especially for these patients options for future therapy are limited. There is therefore, a continued need for new antiretroviral drugs with favorable resistance profiles. Tipranavir, a relatively new protease inhibitor (PI), is active against many strains of HIV-1 that are resistant to several well established PIs, 4 has been approved for the treatment of previously treated adults with HIV-1 infection in the US and Europe. Tipranavir must be administered in combination with low-dose ritonavir (R), which boosts its bioavailability, allowing twice-daily administration. 5
Pharmacodynamic Properties
Tipranavir is a nonpeptidic, dihydropyrone, HIV-1 PI that contains a sulfonamide moiety. 6 As such, its general chemical structure differs from those of other PIs. Tipranavir binds to the active site of HIV-1 protease, 7 thereby preventing the production of mature virions through the supression of protease-based viral processing of gag and gag pol polyproteins. 8 Tipranavir inhibits peptide cleavage by HIV-1 protease with high potency and selectivity in enzymatic assays (inhibition constant 8.9 pmol/L); it is also active against HIV-2 protease (inhibition constant <1 nM). 9
Binding of tipranavir to wild-type HIV-1 protease is mediated by seven direct hydrogen bonds, a water-mediated hydrogen bond and van der Waals interactions. Binding results in a large favorable entropy change (−14.6 kcal/mol) and a small but also favorable enthalpy change (−0.7 kcal/mol at 25 °C). Hydrogen bonding occurs in backbone and catalytic residue regions of the protease that are conserved even in mutant viruses. Evidence suggests that tipranavir maintains significant activity against mutant HIV-1 protease by either compensating for losses in entropy with gains in enthalpy or by limiting losses in enthalpy when binding to mutant protease. The exact binding interactions that contribute to this unique thermodynamic response of remain to be fully elucidated. However, this thermodynamic response is thought to be associated with the high degree of conservation of tipranavir hydrogen bonding sites within mutant viruses as well as the fact that tipranavir binding is less reliant on water-mediated hydrogen bonds compared with other PIs. 10
Antiviral activity
Tipranavir shows good in vitro activity against a broad range of laboratory strains and clinical isolates of HIV-1. Indeed, in vitro, tipranavir effectively inhibits the replication of most HIV-1 group M non-clade B isolates (A, C, D, F, G, H, CRF01 AE, CRF02 AG and CRF12 BF), but it demonstrates less activity against group O isolates. 11 An analysis of a small number of subtype G, CRF02 AG, F and C HIV-1 isolates found tipranavir susceptibility to be the highest in subtype G samples and lowest in subtype F samples. 12 In acute in vitro models of T-cell infection, the 50% effective concentration (EC50) of tipranavir against the replication of clinical isolates and laboratory strains of HIV-1 ranged from 0.03 to 0.07 μmol/L (18 to 42 ng/mL) and the 90% effective concentration (EC90) ranged from 0.07 to 0.18 μmol/L (42 to 108 ng/mL).8,9,11
Against group O isolates, the EC50 ranged from 0.16 to 1 μmol/L and the EC90 from 0.23 to 0.52 μmol/L. As a result of the extensive binding of tipranavir to human plasma proteins, the concentration of tipranavir necessary to effectively inhibit HIV-1 replication in vivo is 3.75-fold higher than the concentration necessary in human plasma-absent experiments. It is therefore estimated that, for the majority of HIV-1 isolates, the in vivo EC90 would range from 0.26 to 0.68 μmol/L (157 to 410 ng/mL). 9
Resistance
Resistance to tipranavir is slow to develop in vitro, and the process is complex and not yet completely understood. However, it is known that the accumulation of a relatively large number of mutations (vs. those associated with most other available PIs) is required to confer resistance, suggesting there is a high genetic barrier to the development of resistance to tipranavir in the clinical setting. In addition, the multiple mutations required to produce marked resistance to tipranavir suggest that it may be useful in the treatment of patients infected with strains of HIV-1 with complex resistance genotypes. 13
During sequential passage of wild-type HIV-1 with increasing concentrations of tipranavir in culture over 9 months (≈70 passages), the first mutant viruses emerged after passage 16 (with tipranavir 800 nmol/L) and had a dominant viral genotype (five of eight viruses) with L33F and I84V mutations in viral protease. However, these viruses did not show an increase in resistance to tipranavir. Although resistance to tipranavir increased with increasing numbers of mutations, ≥10-fold resistance to tipranavir was not conferred until most emergent viruses had six protease mutations. Following passage 33 (with tipranavir 1000 nmol/L), replicating viruses had between two and four mutations and 1.4-fold resistance to tipranavir. Following passage 39 (with tipranavir 2000 nmol/L), replicating viruses had four to six protease mutations (dominant genotyp [15 of 19 viruses]: I13V, V32I, L33F, K45I, I84V) and 10-fold resistance to tipranavir. Viruses emergent after passage 73 (with tipranavir 20 μmol/L) had 87-fold reduced susceptibility to tipranavir. The dominant genotype following this passage had ten protease mutations (L10F, I13V, V32I, L33F, M36I, K45I, I54V, A71V, V82L, I84V) and a V362I mutation in the P2 residue of the polyprotein CA/SP1 cleavage site. Resistance to tipranavir was not increased, nor was viral replication capacity improved, with the mutation in the gag cleavage site. 13
From multiple stepwise regression analyses of clinical studies, 21 mutations in 16 protease positions (10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D and 84V), many of which were identified in the in vitro analysis, have been further associated with reduced susceptibility and lessened virological response to tipranavir. As the number of these mutations increased, the median fold-change in the concentration of tipranavir needed to achieve 50% inhibition (IC50) of the mutant virus compared with the IC50 of wildtype virus also increased. In this analysis, one or no mutations were associated with a <1-fold change in tipranavir susceptibility. Four mutations were associated with a 2-fold change, seven mutations with a 3.9-fold change and eight mutations with a 14.7-fold change. According to predictive regression models, each mutation was also associated with an accumulative decrease in virological response of 0.16 log10 copies/mL following 24 weeks’ treatment with tipranavir. 14
On-treatment genotyping of viral isolates from 276 patients included in clinical trials also indicate that the mutations predominantly emerging with tipranavir treatment are L33F/I/V, V82T/L and I84V. 9 The L33F/I/V, V82T and I84V mutations were present in >20% of HIV-1 isolates from 91 patients included in clinical trials who experienced virological failure on tipranavir/ritonavir treatment. Mutations that were present in 10%-20% of virological failure isolates included L10V/I/S, I13V, E35D/G/N, I47V, K55R, V82L and L89V/M/W. The V82T mutation was present in 34% of virological failure isolates and often developed when there was a V82A mutation at baseline. Isolates with wildtype V82 at baseline more frequently developed the V82L mutation. 15
Preliminary results from another analysis of isolates from patients receiving tipranavir/ritonavir also suggest that mutations in protease codons 36, 58, 69 and 89 are associated with diminished virological response to tipranavir/ritonavir. 16 The replication capacity of HIV-1 in vitro is inverse to the degree of viral resistance to tipranavir. The growth rate of HIV-1 with >3-fold greater tipranavir resistance is <1% of that of wild-type virus in the same conditions. 9 Replicative capacity assays, using Jurkat T-cells containing a luciferase gene driven by an HIV-1 long terminal repeat reporter, showed infection with clones of the dominant virus cultivated after passage 16 (mutations in L33F and I84V regions) gave 55% of the luciferase signal of wild-type virus after 13 days. Clones of the dominant virus emerging after 33-73 passages with tipranavir (three to ten mutations) showed a maximum luciferase signal that was 1% that of wild-type virus at the same timepoint. 14
Mutations of the HIV-1 wild-type virus that confer resistance to tipranavir in vitro also confer resistance to other commonly used PIs with the exception of saquinavir 11 and darunavir (reviewed by Youle). 2 Clones of the dominant viral variant selected after 73 passages with tipranavir (ten protease mutations) had 69-fold greater resistance to tipranavir, 29-fold greater resistance to amprenavir 134-fold greater resistance to atazanavir, 90-fold greater resistance to indinavir, 59-fold greater resistance to lopinavir, 22-fold greater resistance to nelfinavir, 164-fold greater resistance to ritonavir and 3-fold greater resistance to saquinavir. 13 In contrast, the majority of viruses showing resistance to other PIs (e.g. amprenavir, atazanavir, lopinavir and ritonavir) retain an important degree of susceptibility (<4-fold resistance) to tipranavir.2,17 Among highly treatment-experienced patients who have received multiple peptidic PIs, very few (<2.5% of tested isolates) have >10-fold greater resistance to tipranavir. 2
Pharmacokinetic Properties
While not definitively quantified because of low solubility absorption of tipranavir is limited when administered without concomitant ritonavir. Co-administration of ritonavir with most PIs results in inhibition of metabolic pathways (CYP3A4), thereby boosting drug levels of the therapeutic PI. Ritonavir boosted PIs exhibit increased potency compared to non-boosted PIs because they achieve higher plasma trough concentrations (Cmin) and in most cases higher peak plasma concentration (Cmax). 18 Co-administration of ritonavir leads to a 4-fold rise of Cmax, a 48-fold rise of Cmin and a 9 times greater exposure (AUC: area under the curve).17,19 As a result, effective plasma/serum tipranavir concentrations can be achieved with a twice-daily dose administation. Ritonavir has a boosting effect on the plasma concentrations of tipranavir through inhibition of hepatic (and intestinal) cytochrome P450 (CYP) 4A and inhibition of the P-glycoprotein (P-gp) efflux pump. 9 The pharmacokinetic profile of tipranavir does not appear to be appreciably altered in elderly patients, patients with renal impairment or patients with mild hepatic impairment.8,11,20 Despite missing data, tipranavir is contraindicated in patients with moderate or severe hepatic impairment.8,11
Absorption and distribution
In animal studies, absolute bioavailability was approximately 30% after 10 mg/kg oral doses. 7 When tipranavir capsules or oral solution were co-administered with ritonavir at steady-state, there were no clinically significant alterations in Cmax, Cmin, and AUC under fed conditions (500 to 682 kcal, 23 to 25% calories from fat) compared to fasted conditions; so tipranavir may be given with or without food.8,11
The protein binding of tipranavir is greater than 99%.8,11,21 Tipranavir binds to both human serum albumin and alpha-1-acid glycoprotein. In healthy volunteers and HIV-1 positive patients, the mean fraction of tipranavir (dosed without ritonavir) unbound in plasma was similar. Total plasma concentrations ranged from 9 to 82 micromolar. Over this concentration range, the unbound fraction of tipranavir appeared to be independent of total drug concentration.8,11
The volume of distribution in HIV positive patients was 7.7 liters in females (n = 14) and 10.2 liters in males (n = 106) after administration of tipranavir 500 mg/ritonavir 200 mg twice daily for greater than 2 weeks.8,11
In pediatric patient the mean volume of distribution was 4 liters for children 2 to less than 6 years of age (n = 12), 4.7 liters for children 6 to less than 12 years of age (n = 8), and 5.3 liters for children 12 to 18 years of age (n = 6) after administration of tipranavir 375 mg/m2/ritonavir 150 mg/m2.8,11 It is unknown if tipranavir distributes into human sancturary sites like cerebrospinal fluid or semen.8,11
Drug concentration levels
Compared to single doses, peak plasma levels are reduced during multiple-dose administration. This is most likely related to enzyme induction. 22
In HIV infected adults (female: n = 14, males: n = 106) after administration of tipranavir 500 mg/ritonavir 200 mg twice daily for greater than 2 weeks, the mean Cmax was 94.8 ± 22.8 micromolar for females and 77.6 ± 16.6 micromolar for males, the mean Tmax was 2.9 hours for females and 3 hours for males, the mean C trough was 41.6 ± 24.3 micromolar for females and 35.6 ± 16.7 micromolar for males, and the mean AUC was 851 ± 309 micromolar × hr for females and 710 ± 207 micromolar × hr for males.8,11
In pediatric patients after administration of tipranavir 375 mg/m2/ritonavir 150 mg/m2 the mean Cmax was 135 ± 44 micromolar for children 2 to less than 6 years of age (n = 12), 151 ± 32 micromolar for children 6 to less than 12 years of age (n = 8), and 138 ± 52 micromolar for children 12 to 18 years of age (n = 6), the mean Tmax was 2.5 hours for children 2 to less than 6 years of age (n = 12), 2.6 hours for children 6 to less than 12 years of age (n = 8), and 2.7 hours for children 12 to 18 years of age (n = 6), the mean C trough was 59.6 ± 23.6 micromolar for children 2 to less than 6 years of age (n = 12), 66.3 ± 12.5 micromolar for children 6 to less than 12 years of age (n = 8), and 53.3 ± 32.4 micromolar for children 12 to 18 years of age (n = 6), and the mean AUC was 1190 ± 332 micromolar × hr for children 2 to less than 6 years of age (n = 12), 1354 ± 256 micromolar × hr for children 6 to less than 12 years of age (n = 8), and 1194 ± 517 micromolar × hr for children 12 to 18 years of age (n = 6).8,11
Steady-state plasma trough concentrations at 10 to 14 hours from clinical trials showed that females generally had higher tipranavir concentrations than males. The median plasma trough concentration of tipranavir was 43.9 micromolar for females (n = 14) and 31.1 micromolar for males (n = 106) after 4 weeks of tipranavir 500 mg/ritonavir 200 mg twice daily dosing. The difference in concentration is not significant enough to require a dosage adjustment.8,11
Metabolism and elimination
Tipranavir is a substrate and inducer of cytochrome P450-3A. The net effect of tipranavir co-administered with ritonavir 200 milligrams is CYP3A4 inhibition. Therefore, drugs highly dependent on CYP3A4 for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening events should be contraindicated.8,11,23
A phenotypic study examining the impact of tipranavir 500 mg/ritonavir 200 mg twice daily on hepatic enzymes at first dose and at steady state was conducted in 16 healthy volunteers. Subjects were administered tipranavir/ritonavir for 10 days. No net effect on CYP2C9 and hepatic P-glycoprotein was seen at first dose or steady state. No net effect was seen after first dose at CYP1A2, however moderate induction was seen at steady state. Modest CYP2C19 inhibition was seen at first dose, however marked induction was seen at steady state. Potent inhibition of CYP2D6 and hepatic and gastrointestinal CYP3A4/5 were seen at first dose and steady state.8,11
The impact of tipranavir/ritonavir on hepatic and gastrointestinal P-glycoprotein was assessed using oral and intravenous digoxin. Study results suggested that P-glycoprotein is inhibited after the first dose, however, at steady state, induction is seen. As a result, it is difficult to predict the impact of tipranavir/ritonavir on drugs that are dual substrates of CYP3A and P-glycoprotein.8,11
In HIV positive adults the mean half-life was 5.5 hours in female patients (n = 14) and 6 hours in males (n = 106) after administration of tipranavir 500 mg/ritonavir 200 mg twice daily for greater than 2 weeks.8,11 In pediatric patients the mean half-life was 8.1 hours for children 2 to less than 6 years of age (n = 12), 7.1 hours for children 6 to less than 12 years of age (n = 8), and 5.2 hours for children 12 to 18 years of age (n = 6) after administration of tipranavir 375 mg/m2/ritonavir 150 mg/m2.8,11
When administered with ritonavir, the majority of tipranavir is excreted unchanged (feces: 79.1%; urine: 3.9%). The most abundant (3.2% of dose) metabolite found in the feces is a hydroxyl metabolite. A glucuronide conjugate metabolite (0.5% of dose) was in the urine.8,11
Tipranavir is highly protein bound, and dialysis is not expected to remove a significant amount of the drug.8,11
Drug interactions
General drug interactions
Tipranavir is a substrate and inducer of cytochrome CYP3A. The net effect of tipranavir co-administered with ritonavir 200 milligrams is CYP3A4 inhibition. Therefore, drugs highly dependent on CYP3A4 for clearance or are potent CYP3A inducers (e.g. antiarrhythmics (amiodarone, bepridil, quinidine, as well as flecainide propafenone (which rely on CYP2D6 for clearance)), antihistamines (astemizole, terfenadine), ergot derivatives (dihydroergotamine, ergonovine, ergotamine, methylergonovine), gastromotility agents (cisapride), neuroepileptics (pimozide), sedatives/hypnotics (triazolam) and phytotherapeutics (St. John's wort, garlic) should not be administered with tipranavir.8,11
In vitro, tipranavir also inhibits the enzymes CYP1A2, CYP2C9, CYP2C19 and CYP2D6.8,11 Given that ritonavir is also a CYP2D6 inhibitor, the net effect of tipranavir/ritonavir on this enzyme may be inhibition. In vivo, the overall effect of tipranavir/ritonavir on CYP1A2, CYP2C9 and CYP2C19 is not known. There are no data available regarding any induction effect of tipranavir on these enzymes, or any inhibitory or induction effect of tipranavir on glycuronosyl transferases.8,11 Therefore, as with other PIs 24 complexity characterises the drug interaction profile of tipranavir/ritonavir and attention must be given to the co-administration of tipranavir/ritonavir with other pharmacotherapies.8,11
Interaction with antiretroviral drugs
The majority of nucleoside and nucleotide reverse transcriptase inhibitors (NRTIs) do not affect the CYP enzyme system to any significant extent and dosage adjustments of tipranavir are not generally required when it is coadministered with these agents. 11 However, reductions in systemic exposure to abacavir and zidovudine have been observed when they are coadministered with tipranavir, concomitant administration of tipranavir and abacavir or zidovudine is not generally recommended unless no other NRTIs are available and suitable. 11 Although no clinically significant change in the pharmacokinetic profile of didanosine was observed in healthy volunteers who received tipranavir/ritonavir 500 mg/100 mg or 750 mg/200 mg with a single dose of the enteric-coated formulation of didanosine 400 mg in one study, 25 a reduction in the AUC of didanosine when these two drugs are coadministered has been reported. 11 However, the clinical relevance of this reduction has not been determined.8,11 Tipranavir administered as a soft play formulation incompatability when coadministered and therefore should be administered with an interval of ≥2 hours.8,11
There do not appear to be any clinically significant pharmacokinetic interactions between tipranavir and the non-nucleoside reverse transcriptase inhibitors (NNRTIs) efavirenz 26 and nevirapine. 11 In healthy volunteers systemic exposure to efavirenz during coadminstration with tipranavir/ritonavir was not markedly different from that in historical controls. 26
The pharmacokinetic studies suggest that tipranavir and other PIs should not be combined as a result of marked decreases in plasma concentrations of the latter (amprenavir, lopinavir, saquinavir minimum concentrations [Cmin] decreased 51%, 45% and 84%, respectively). 27
Preliminary data also suggest a possible drug interaction between enfuvirtide and tipranavir/ritonavir resulting in higher trough concentrations of tipranavir and ritonavir.28,29 However, this has not been reported to be clinically relevant in terms of hepatotoxicity. 29
As tipranavir/ritonavir reduces systemic exposure to etravirine, it has been suggested that coadministration should be avoided.2,30
A reduction in systemic exposure to raltegravir when administered with tipranavir/ritonavir has also been reported, 31 although it has been suggested that these agents may be coadministered without dosage modification. 32 Low trough levels of tipranavir are reported in a combination antiretroviral therapy with tenofovir. 33
Therapeutic Efficacy
Tipranavir, co-administered with ritonavir and in conjunction with other antiretroviral agents, is indicated for the treatment of HIV-1 infected adults and pediatric patients greater than 2 years of age who have evidence of viral replication and are highly treatment-experienced or have HIV-1 strains resistant to multiple protease inhibitors. Low-dose ritonavir is required with tipranavir to achieve higher and more sustained tipranavir plasma levels. The indication was based on 2 controlled studies of 48 weeks duration in adults and one controlled study of 48 weeks duration in pediatric patients 2 to 18 years of age that demonstrated improvement in plasma HIV-1 RNA levels. The treatment population for both adult studies was clinically advanced, 3-class antiretroviral (NRTIs, NNRTIs, and PIs) treatment-experienced adults with evidence of HIV-1 replication despite ongoing antiretroviral therapy. There are no study results demonstrating the effect on clinical progression of HIV-1. Virologic response is dependent on the number of baseline primary protease inhibitor mutations. Major limitations of tipranavir/ritonavir therapy is the risk of hepatitis and hepatic decompensation including fatalities and serious drug interactions. The risk-benefit of tipranavir has not been established in treatment-naïve adult and pediatric patients or in pediatric patients less than 2 years of age.8,11
The RESIST (Randomized Evaluation of Strategic Interventions in Mulitdrug Resistant Patients with Tipranavir) trials
Data combined from 2 randomized, controlled, open-label, multicenter studies (n = 1509) demonstrate that at 48 weeks, tipranavir/ritonavir produced better virologic and immunologic response than other protease inhibitors/ritonavir in adults with evidence of viral replication, who are highly treatment-experienced or have HIV-1 strains resistant to multiple protease inhibitors compared with other protease inhibitors. Patients were randomized to either tipranavir 500 mg plus ritonavir (TPV-ritonavir) 200 mg twice daily or a comparator protease inhibitor plus ritonavir (CPI-ritonavir) twice daily. The CPI-ritonavir twice daily regimens included lopinavir 400 mg/ritonavir 100 mg, amprenavir 600 mg/ritonavir 100 mg, saquinavir 800 mg/ritonavir 200 mg or 1000 mg/ritonavir 100 mg, or indinavir 800 mg/ritonavir 100 mg. Both groups were also on patient-specific optimized background regimens based on each patient's genotypic resistance testing and patient history. At baseline, patients had extensive prior exposure to a median of 6 nucleoside reverse transcriptase inhibitors, 1 non-nucleoside reverse transcriptase inhibitors and 4 protease inhibitors. In addition, 10% of patients also had previous exposure to enfuvirtide. Median baseline plasma HIV-1 RNA was 4.73 log 10 copies/mL and median baseline CD4+ cell count was 196 cells/microliter. Primary efficacy endpoints for both studies were proportion of treatment responders at 48 weeks and time to treatment failure. Of the 1509 patients originally randomized, 26 did not receive treatment, therefore, the pooled efficacy population consisted of 1483 patients. By week 48, 65.1% of tipranavir-ritonavir patients and 26.1% of CPI-ritonavir patients completed the 48-week visit and were analyzed. The proportion of virological responders (confirmed at least 1 log 10 HIV-1 RNA below baseline) was 33.6% for TPV-ritonavir and 15.3% for CPI/ritonavir (p less than 0.0001) in a pooled analysis of the 2 studies at 48 weeks. In both treatment groups, a greater proportion of patients receiving enfuvirtide in the optimized background regimen responded to treatment than those not receiving enfuvirtide (p less than 0.001). Through the 48 weeks of treatment, the proportion of patients with HIV-1 RNA less than 400 copies/mL were 30.4% and 13.8% in the TPV-ritonavir and CPI/ritonavir groups, respectively (p less than 0.0001) and with HIV-1 RNA less than 50 copies/mL were 22.8% and 10.2%, respectively (p less than 0.0001). The mean change in baseline CD4 cell count at the last measurement up to week 48 was +32 cells/microliter in the TPV-ritonavir group and +18 in the CPI-ritonavir group (p less than 0.0001). When enfuvirtide was part of the optimized background regimen, the mean increase in CD4 count was 88 and 33 cells/microliter in the TPV-ritonavir and CPI-ritonavir groups, respectively. TPV-ritonavir was also associated with a greater mean reduction in viral load compared with CPI-ritonavir (−1.14 log 10 copies/mL and −0.54 log 10 copies/mL, respectively; p less than 0.0001). The median time to treatment failure was 113 days in the TPV-ritonavir group compared to 0 days in the CPI-ritonavir group (p less than 0.0001). The median time to treatment failure nearly tripled in the TPV-ritonavir group when enfuvirtide was used (p = 0.0002), but it did not prolong the median time to treatment failure in the CPI-ritonavir group. Overall, the adverse event profile between the two treatment groups was similar with mild gastrointestinal events being reported most often. 4
Efficacy, safety and tolerability of tipranavir coadministered with ritonavir in HIV-1-infected children and adolescents
In treatment-experienced HIV-1 infected pediatric patients (n = 110) treated with high- or low-dose tipranavir plus ritonavir in conjunction with an optimized background antiretroviral regimen, the regimen provided a sustained virologic response (secondary endpoint), and was found to be safe and well-tolerated (primary endpoint) according to an ongoing, 48-week, randomized, open-label, phase I/IIa study. The study included HIV-1 infected, treatment-experienced pediatric patients (with the exception of 3 treatment-naive patients), with baseline HIV-1 RNA of at least 1500 copies/mL. Resistance to all commercially available protease inhibitors was present in 49.6% (n = 57) of patients at baseline. Patients were randomized to receive either high-dose tipranavir/ritonavir 375 milligrams/square meter (mg/m2)/150 mg/m2 twice daily (n = 57), or low-dose tipranavir/ritonavir 290 mg/m2/115 mg/m2 twice daily (n = 58) with additional therapy of at least two non-protease inhibitor antiretroviral drugs, optimized using baseline genotype resistance testing. Tipranavir was available as an oral solution or soft-gel capsule, with a maximum tipranavir/ritonavir dose of 500 mg/200 mg twice daily. Patients age 2 to 18 years included in the study had a median baseline plasma HIV-1 RNA of 4.7 log(10) copies/mL and a median baseline CD4+ cell count of 378.5 cells/microliter, which were comparable between the 2 treatment groups. The 48-week study was completed by 88 (76.5%) patients. The rate of discontinuations primarily due to virologic failure was not significantly different between the 2 treatment groups. A virologic response was defined as patients achieving and maintaining a viral load of less than 400 copies/mL, less than 50 copies/mL, and a viral load reduction of at least 1 log(10) copies/mL. Although the study was not powered for efficacy, the proportion of patients with a viral load less than 400 copies/mL increased from baseline until week 8 and then stabilized until week 48. A viral load of less than 400 copies/mL occurred at week 48 in 45.6% (25/57) and 39.7% (23/58) (p = 0.57) of patients in the high-and low-dose groups, and less than 50 copies/mL in 35.1% vs. 34.5% (p = 0.84) of patients, respectively. The greatest virologic response when defined as viral load of less than 400 copies/mL occurred in patients 2 to less than 6 years of age as shown in Table 1.
Virologic response (less than 400 copies/ml) at 48 weeks by age groups.34

The median HIV RNA count reductions from baseline were not significantly different between groups, −0.8 log(10) vs. −1.24 log(10) in the low- and high-dose groups. Four patients, all in the low-dose group, developed AIDS-defining illness through week 48. The safety and tolerability of the low- and high-dose treatment regimens were not significantly different with serious adverse events occurring in 27.6% (16/58) vs. 22.8% (13/57), respectively. 34
Tolerability
In the RESIST-trials, adverse events with tipranavir/ritonavir were typical of those seen with the ritonavir-boosted CPIs, despite the higher dosage of ritonavir received by patients in the tipranavir/ritonavir group. Of note, it has been reported that ritonavir trough concentrations in recipients of tipranavir/ritonavir are lower than trough concentrations achieved when lower doses are administered with saquinavir or lopinavir, which may be due to differences in the effects of these drugs on the metabolism of ritonavir. 4
In the RESIST-trials, moderate to severe diarrhea was reported in 15% of patients receiving tipranavir plus ritonavir 500 mg/200 mg twice daily (n = 749) compared with 13.4% of patients receiving a comparator protease inhibitor plus ritonavir (n = 737), and was one of the most common adverse effects reported. Comparator protease inhibitors included lopinavir/ritonavir (400 mg/100 mg twice daily), indinavir/ritonavir (800 mg/100 mg twice daily), saquinavir/ritonavir (1000 mg/100 mg twice daily), and amprenavir/ritonavir (600 mg/100 mg twice daily).
Hyperlipidemia was reported in 2.5% of patients receiving tipranavir plus ritonavir 500 mg/200 mg twice daily (n = 749) compared with 0.8% of patients receiving a comparator protease inhibitor plus ritonavir (n = 737). Large increases in total cholesterol and triglycerides have occurred in patients on tipranavir/ritonavir 500 mg/200 mg twice daily (Table 2).
Lipide profile during RESIST-trials.4
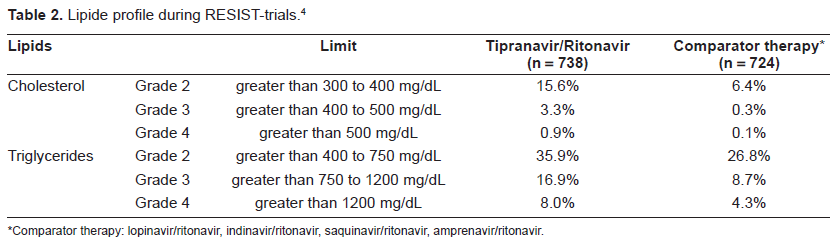
Comparator therapy: lopinavir/ritonavir, indinavir/ritonavir, saquinavir/ritonavir, amprenavir/ritonavir.
Clinical hepatitis and hepatic decompensation, including some fatalities, have been associated with tipranavir/ritonavir administration (Table 3). Causal relationship could not be established. Patients at greatest risk are patients with advanced HIV disease taking multiple concomitant medications and/or patients with chronic hepatitis B or C coinfection. Patients with chronic hepatitis B or C coinfection or elevations in transaminases are at an approximately 2-fold greater risk for developing grade 3 or 4 transaminase elevations or hepatic decompensation. Liver function should be monitored, and tipranavir/ritonavir should be discontinued if patients develop signs or symptoms of hepatitis such as fatigue, malaise, anorexia, nausea, jaundice, bilirubinuria, acholic stools, liver tenderness, or hepatomegaly.8,11
Transaminase elevation during RESIST-trials.4
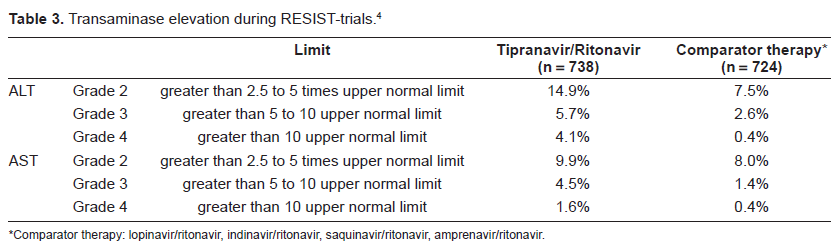
Comparator therapy: lopinavir/ritonavir, indinavir/ritonavir, saquinavir/ritonavir, amprenavir/ritonavir.
During clinical trials, 14 intracranial hemorrhage events were reported in 6,840 HIV-1 infected individuals treated with tipranavir/ritonavir in combination with other antiretroviral therapy. There were 8 fatal cases. Most of the patients had comorbidities such as CNS lesions, head trauma, recent neurosurgery, coagulopathy, hypertension, or alcohol abuse or were receiving concomitant medications such as anticoagulants and antiplatelet drugs, which may have contributed to the hemorrhage. The median time to onset of an intracranial hemorrhage event and initiation of tipranavir/ritonavir therapy was 525 days. Most patients had no evidence of abnormal coagulation during or prior to the event. Therefore, routine monitoring of coagulation parameters is not recommended. Inhibition of platelet aggregation were similar between tipranavir and tipranavir/ritonavir in in vitro experiments.8,11 In a case report of a patient on a HAART regimen of ritonavir boosted tipranavir, the patient developed an intracranial hemorrhage during an acute phase of cryptococcal meningitis. 35
Dosage and administation
Tipranavir is approved in HIV-1 infected pediatric patients greater than 2 years of age and adult patients who are treatment-experienced and have HIV-1 strains resistant to multiple protease inhibitors; safety and effectiveness have not been established in pediatric patients less than 2 years of age.8,11
The recommended dose for pediatric patients 2 years and older is tipranavir 14 mg/kg orally co-administered with ritonavir 6 mg/kg orally (or tipranavir 375 mg/m2 orally co-administered with ritonavir 150 mg/m2 orally) given twice daily in combination with other antiretroviral treatment. The dose should not exceed a maximum dose of tipranavir 500 mg co-administered with ritonavir 200 mg twice daily. For pediatric patients who develop intolerance or toxicity and cannot continue tipranavir 14 mmg/kg with 6 mg/kg ritonavir orally twice daily, a dose reduction to tipranavir 12 mg/kg with ritonavir 5 mg/kg (or tipranavir 290 mg/m2 co-administered with 115 mg/m2 ritonavir) orally twice daily may be considered. A dose reduction is not appropriate for patients whose virus is resistant to multiple protease inhibitors.
The recommended oral dose for aduld patients is tipranavir 500 mg, co-administered with ritonavir 200 mg, twice daily in combination with other antiretroviral treatment. The formulation of tipranavir is 250 mg per pill and for ritonavir 100 mg per pill, which means a pill burden of eight pills per day for one single PI. Tipranavir and ritonavir can be taken with or without food. No dose adjustments are necessary for males or females There are no pharmacokinetic data for tipranavir in patients with renal impairment. Renal clearance of tipranavir is negligible, however, and a decrease in total body clearance is not expected. Tipranavir/ritonavir is contraindicated in patients with moderate or severe hepatic impairment (Child-Pugh Class B or C, respectively). No dose adjustment is necessary for patients with mild hepatic impairment. Discontinue tipranavir/ritonavir in patients who develop asymptomatic elevations in aspartate transaminase (AST) or alanine aminotransferase (ALT) greater than 10 times the upper limit of normal or in patients with asymptomatic elevations in AST or ALT between 5 to 10 times the upper limit of normal with increases in total bilirubin greater than 2.5 times the upper limit of normal.
Place of Tipranavir in the Management of HIV-1 Infection
Antiretroviral therapy (ART) remains the mainstay of HIV-1 treatment. Ritonavir-boost PIs are a standard-of-care firstline therapy for the management of HIV-1. Unfortunately, however, even with ART antiretroviral treatment failure is common.
Specific data pertaining to the prevalence of patients who have extensive antiretroviral experience and resistance to multiple antiretroviral drugs are limited. 3 However, among the 8500 patients included across Europe in the EuroSIDA cohort, 413 (5%) were reported to have had both treatment with antiretroviral drugs from all three main drug classes and virological failure with two HAART regimens. 36 It is also estimated that around 40% of patients receiving HIV-1 care in the US with viral loads > 500 copies/mL have resistance to one or more PIs and ã13% have resistance to all three drug classes. 37
Treatment guidelines acknowledge the complexity of effectively managing patients with current treatment failure and extensive prior antiretroviral experience.1,24,38 It is recommended that evaluation of such patients include drug resistance testing as well as assessment of both the severity of HIV-1 disease and antiretroviral treatment history, the latter taking into account the duration of prior treatment, previous drugs used and their potency, adherence history and drug intolerance and toxicity. The aim of treatment for previously treated patients showing drug resistance is the achievement of maximal suppression of HIV RNA levels to <50 copies/mL. However, where this is unachievable, treatment should aim to preserve immune function and prevent disease progression. ART usually consists of at least three antiretroviral drugs, generally selected from the NRTI, NNRTI and PI classes of drugs.
When boosted with ritonavir, tipranavir (like darunavir, another new ritonavir-boosted PI) is used as a component of ART regimens in the management of treatment-experienced patients who are infected with HIV-1 strains resistant to multiple PIs, in keeping with its activity in vitro. Tipranavir/ritonavir is approved for use in treatment-experienced patients in both Europe 11 and the US. 8 In Europe, tipranavir/ritonavir is specifically indicated for extensively treatment-experienced patients who harbour HIV-1 strains resistant to multiple PIs. US prescribing information recommends tipranavir, coadministered with ritonavir, as combination treatment for treatment-experienced patients infected with HIV-1 strains resistant to more than one PI.
These approved indications are supported by data from two well designed trials (RESIST-1 and −2) that demonstrated that twice-daily tipranavir/ritonavir 500 mg/200 mg was more effective than ritonavir-boosted CPI (amprenavir, indinavir, lopinavir or saquinavir) treatment in terms of sustained treatment response and length of time to treatment failure when both treatments were given along with an optimized background regimen in heavily antiretroviral-experienced patients. Patients included in the trials had genotypically demonstrated resistance to other PIs (section 4). In these studies, predictors of effective treatment response to tipranavir/ritonavir were (i) use of enfuvirtide as part of of the optimized background regimen; (ii) fewer mutations considered to be associated with decreased tipranavir activity at baseline; and (iii) higher trough concentrations of tipranavir. Additionally, patients with at least two primary PI-associated mutations at baseline and those who had previously received treatment with at least three PIs were significantly more likely to have a beneficial treatment response with tipranavir/ritonavir treatment than patients receiving CPI/ritonavir treatment (section 4). Recipients of tipranavir/ritonavir with a viral load reduction of ≥1.5 log10 copies/mL at week 8 were more likely to achieve an undetectable viral load at week 48. Recent evidence indicates that the efficacy of tipranavir/ritonavir is sustained for up to 3 years (section 4).
The approval of tipranavir for patients with viral resistance to other PIs is also supported by the pharmacodynamic properties of the drug. Tipranavir appears to maintain binding efficacy for mutant HIV-1 protease strains through a unique thermodynamic response, and the majority of viruses resistant to other approved PIs remain susceptible to tipranavir (section 2). Importantly, resistance to tipranavir is slow to develop and the accumulation of a relatively large number of mutations (vs. those associated with most other available PIs) is required to confer resistance, suggesting that there is a high genetic barrier to the development of resistance to tipranavir in the clinical setting. In addition, the multiple mutations required to produce marked resistance to tipranavir suggest that it may be useful in the treatment of patients infected with strains of HIV-1 with complex resistance genotypes. 13
However, while well designed trials have provided evidence that the promising in vitro pharmacodynamic properties of tipranavir translate into important clinical benefit when administered to heavily treatment-experienced patients in combination with low-dose ritonavir, use of tipranavir/ritonavir is limited by the drug interaction profile of the combination. Particularly associated with the effects of tipranavir/ritonavir on CYP isoenzymes and P-gp, tipranavir/ritonavir has been shown to, or is expected to, interact to a clinically significant effect with a diverse array of other pharmacotherapies (section 3).
Some important tolerability issues must also be taken into account when considering the use of tipranavir/ritonavir. While there were no reports of intracranial hemorrhage in the pivotal RESIST trials (section 4), across all clinical trials of tipranavir a small number of intracranial hemorhages were observed (section 5). It is therefore recommended that tipranavir be administered with caution in patients with an increased bleeding risk (section 5). Because of the association with the risk of clinical hepatitis, prescribing information recommends that patients receiving tipranavir/ritonavir who have an increased risk of hepatotoxicity, such as those with chronic HBV or HCV co-infection, be closely monitored. Risk/benefit assessments are also vital in any decision to use tipranavir in this patient population. In the RESIST trials (section 4), elevated transaminases were significantly more frequent in tipranavir/ritonavir-treated patients than patients receiving a CPI/ritonavir (section 5).
An additional important tolerability issue highlighted by RESIST data is the increased risk of dyslipidemia with tipranavir/ritonavir treatment compared with other PIs (section 5).
Of note, regarding the implications of the tolerability profile of tipranavir/ritonavir, health-related quality of life (HR-QOL) data (assessed by use of the Medical MedianOutcomes Study HIV Health Survey) from 984 paincluded in the RESIST trials (see section 4) have been published. 39 The study showed that, despite the incidence of treatment-related adverse HR-QOL was maintained across all summary and subscale scores in the two treatment groups over the 48-week evaluation period. Compared with CPI/ritonavir group, recipients of tipranavir/ritonavir had a small but significant (p < 0.05) increase (4.8 points) in pain scores. Further data on HR-QOL with tipranavir/ritonavir treatment versus other PIs would be of interest.
A direct comparison of HR-QOL with tipranavir/ritonavir versus darunavir/ritonavir would also be particularly useful, as would data on the relative efficacy of the two drugs.
Preliminary evidence also suggests that mutations conferring resistance to tipranavir do not appear to confer resistance to darunavir. 40 This may be relevant in terms of sequencing of these next generation PIs.
Treatment guidelines recommend the inclusion of several active agents in a treatment regimen for treatment-experienced patients with few treatment options (guided by HIV-1 resistance testing). Combined results from the RESIST studies support this recommendation through the demonstration of an important efficacy advantage when enfuvirtide was included as part of the optimized background regimen. Across treatment groups, significantly more patients receiving enfuvirtide achieved and maintained a treatment response to week 48 compared with patients who did not receive enfuvirtide. Median time to treatment failure was also significantly longer across treatment groups among patients who received enfuvirtide versus those who did not and secondary efficacy variables also showed significant differences in favour of enfuvirtide treatment versus no enfuvirtide treatment (section 4). Additionally, among patients who were naïve to enfuvirtide but received it as part of their background regimen during the study, significantly more patients in the tipranavir/ritonavir group had a treatment response than those in the CPI/ritonavir group.
Treatment guidelines also acknowledge that antiretroviral drugs may be candidates for management through therapeutic drug monitoring (TDM). In June 2007, the manufacturer of tipranavir announced the commencement of the SPRING (Safety, efficacy and Pharmacokinetics of tipRanavir IN 400 racially and Gender diverse HIV-1-positive, treatment-experienced population) study. 41 The study will investigate the impact of TDM on the efficacy and safety of tipranavir/ritonavir.
Although there is good clinical and pharmacological evidence supporting the use of tipranavir in treatment-experienced patients with HIV-1 infection, other new agents are available that may also be selected for use as components of HAART regimens for previously-treated patients with HIV-1 infection. For example, darunavir, raltegravir, maraviroc and etravirine can now be prescribed in some countries and may be selected by the clinician on the basis of their efficacy, tolerability and resistance profiles. The efficacy of raltegravir is currently being evaluated in combination with tipranavir and other agents in patients with triple class-resistant virus in two large, randomized, doubleblind trials.
In conclusion, the durable efficacy of tipranavir, in combination with low-dose ritonavir (tipranavir/ritonavir 500 mg/200 mg twice daily), has been demonstrated in well designed trials in treatment-experienced adults infected with multidrug-resistant HIV-1. In treatment-experienced patients with HIV-1 infection receiving an optimized background regimen, viral suppression was greater and immunological responses were better with regimens containing tipranavir/ritonavir than with other ritonavir-boosted PI-containing regimens. The efficacy benefit appeared to be more marked in patients receiving two fully active drugs in the regimen, with the combination of tipranavir/ritonavir and enfuvirtide (for the first time) appearing to be the most successful. Although tipranavir is generally well tolerated, clinical hepatitis and hepatic decompensation, and intracranial hemorrhage have been associated with the drug. Tipranavir also has a complex drug interaction profile. Thus, tipranavir, administered with ritonavir, is an effective treatment option for use in the combination therapy of adults with HIV-1 infection who have been previously treated with other antiretroviral drugs.
Summary
Pharmacological properties
Tipranavir is a highly potent and selective nonpeptidic sulfonamide-containing dihydropyrone HIV-1 PI with good in vitro activity against a broad range of laboratory strains and clinical isolates of HIV-1. In vitro, resistance to tipranavir is slow to develop and the process is complex. On-treatment genotyping of viral isolates from treatment-experienced patients in clinical trials indicate that the mutations predominantly emerging with tipranavir treatment are L33F/I/V, V82T/L and I84V. In vitro, mutations of the HIV-1 wild-type virus that confer resistance to tipranavir also confer resistance to most other commonly used PIs, with the exception of saquinavir and darunavir. The activity of tipranavir is reduced in the presence of human plasma proteins. Synergistic, additive and antagonistic effects have been observed with tipranavir and various other antiretroviral drugs in vitro. Tipranavir is administered in combination with low-dose ritonavir (tipranavir/ritonavir), which boosts its bioavailability resulting in the achievement of effective plasma/serum concentrations of tipranavir with a twice-daily regimen. Depending on the dose of tipranavir/ritonavir administered, maximum plasma tipranavir concentrations are reached within 1-5 hours and, in the majority of individuals, steady state is reached after approximately 7 days of twice-daily administration. At steady state, tipranavir pharmacokinetics are linear when tipranavir is boosted by low-dose ritonavir. The effect of tipranavir/ritonavir on cytochrome P450 (CYP) isoenzymes, particularly CYP3A, and on P-glycoprotein (P-gp, ABCB1) results in numerous pharmacokinetic interactions between tipranavir/ritonavir and other drugs. At steady state, tipranavir/ritonavir shows moderate inhibitory effects on hepatic CYP3A4/5, strong inhibitory effects on intestinal CYP3A4/5 and a minimal effect on the activity of P-gp, according to preliminary results of a well designed drug interaction study in healthy volunteers.
Therapeutic efficacy
The therapeutic efficacy of oral tipranavir/ritonavir 500 mg/200 mg twice daily versus that of a ritonavir-boosted comparator PI (CPI) [lopinavir, indinavir, saquinavir or amprenavir] has been evaluated in the similarly designed randomized, nonblind, multicenter RESIST (Randomized Evaluation of Strategic Intervention in multidrug reSistant patients with Tipranavir)-1 and RESIST-2 trials in highly antiretroviral-experienced, HIV-1-infected adult patients. All patients also received concurrent treatment with an optimized background regimen (with or without enfuvirtide).
Study participants were HIV-1-infected adult patients who had received ≥3 months of previous treatment with a nucleoside reverse transcriptase inhibitor, a non-nucleoside reverse transcriptase inhibitor and a PI. Included patients had also previously received at least two PI-based regimens. At baseline, all patients had a plasma HIV-1 RNA level of ≥1000 copies/mL.
Recipients of tipranavir/ritonavir experienced a sustained response to treatment, with a significantly larger proportion of patients achieving a confirmed viral load reduction of ≥1 log10 copies/mL at 48 weeks than recipients of CPIs (primary endpoint; pooled data). In addition, the tipranavir/ritonavir treatment group had a longer time to treatment failure than did patients treated with the CPIs (primary endpoint). Immunological responses, a secondary efficacy measure, were also significantly better in the tipranavir/ritonavir group than in the recipients of CPIs. Preliminary data indicate that the efficacy of tipranavir/ritonavir is sustained for up to 156 weeks.
In pediatric patients (2-18 years) the efficacy, safety and tolerability of ritonavir-boosted tipranavir (TPV/r) has been studied in an open-label randomized pediatric trial (1182.14/PACTG1051) comparing TPV/r at two doses including an optimized background regimen. HIV-1-infected patients (2-18 years) with plasma viral load 1500 copies/ml or more were randomized to TPV/r 290/115 or 375/150 mg/m2 twice-daily oral solution and optimized background regimen. Of one-hundred and fifteen children randomized to low or high dose therapy, eighty-eight remained on-treatment through 48 weeks. At 48 weeks, 39.7% low-dose and 45.6% high-dose TPV/r recipients had viral load less than 400 copies/ml and 34.5 and 35.1%, respectively, achieved viral load less than 50 copies/ml. This means that TPV/r achieved a sustained virologic response, showed a good safety profile and was well tolerated at either dose.
Tolerability
In the RESIST trials, adverse events with tipranavir/ritonavir were typical of those seen with the ritonavir-boosted CPIs, despite the higher dosage of ritonavir received by patients in the tipranavir/ritonavir group. Gastrointestinal adverse events were the most common treatment-emergent events with tipranavir/ritonavir and were usually mild. The adverse event rate per 100 patient-exposure years of any adverse event was similar among patients receiving tipranavir/ritonavir and those receiving a CPI/ritonavir. Exposure-adjusted total rates of grade 3 or 4 laboratory abnormalities were also similar in the the two treatment groups. Grade 3 or 4 elevations in triglyceride levels, cholesterol levels and liver enzymes occurred significantly more frequently among patients receiving tipranavir/ritonavir than in the CPI/ritonavir recipients. However, the most highly treatment experienced patients did not experience grade 3 or 4 AST or ALT elevations during 96 weeks of treatment with tipranavir/ritonavir in five phase IIb/III trials. When reported, grade 3 or 4 AST or ALT elevations were asymptomatic in the majority of patients.
Reports of clinical hepatitis and hepatic decompensation, including some fatalities, have been associated with tipranavir/ritonavir treatment, usually in patients receiving multiple medications and with advanced HIV-1-infection. Caution is recommended and increased liver enzyme monitoring should be considered in patients with liver enzyme abnormalities or hepatitis receiving tipranavir. In addition, tipranavir has been associated with fatal and nonfatal intracranial hemorrhage and should be administered with caution in patients with an increased bleeding risk (see boxed warning in the manufacturer's prescribing information). However, rates of intracranial hemorrage in a large cohort of US Medi-Cal recipients not taking tipranavir were similar to those observed in premarketing trials of tipranavir. Rashes have been reported in patients receiving tipranavir/ritonavir.
Disclosure
The authors report no conflicts of interest.