
Abstract
The mTOR (mammalian target of rapamycin) signaling pathway was discovered during studies of the immunosuppressive agent rapamycin. This pathway regulates cell growth, protein synthesis and angiogenesis in response to environmental factors. The mTOR complex-1 inhibitor temsirolimus was derived from rapamycin to have less immunosuppressive and improved solubility characteristics. The safety, tolerability and efficacy of temsirolimus have been well established in clinical trials. Drug related toxicity included rash, mucositis, asthenia, nausea, hyperglycemia, hypophosphatemia, anemia, and hypertriglyceridemia. An active and well-tolerated single agent dose is 25 mg i.v. weekly. A large Phase III trial in poor-prognosis patients with metastatic renal cancer compared i.v weekly temsirolimus administration to subcutaneous interferon alpha (IFNα), or a combination of temsirolimus plus IFNα. This study established that median overall survival was improved to 10.9 months in the temsirolimus group compared to 7.3 months in IFNα-treated group (0.73 hazard ratio for death; 95% confidence interval [CI], 0.58 to 0.92; P = 0.008). A modest objective response rate of 8.6%, 4.8%, and 8.1%, respectively was observed in the three groups, associated with a median time to treatment failure of 3.8 months for temsirolimus alone, 1.9 months for IFNα, and 2.5 months for the combination. These results led to approval of temsirolimus for the treatment of renal cancer in the United States. Temsirolimus is clearly indicated for first-line therapy of Motzer “poor risk” renal cancer and aggressive non-clear cell renal cancer. Temsirolimus may be useful after failure of VEGF tyrosine kinase inhibitors. Clinical activity in other tumor types, such as endometrial cancer has been observed. Temsirolimus is therefore an important new agent for cancer treatment.
Background
Renal carcinoma (RC) is the most common malignancy arising from the kidney, accounting for 3% of all adult cancers. In the United States, it is estimated that 54,390 new cases of renal cancer (including cancers of the renal pelvis), and 13,010 deaths due to metastatic disease occurred in 2008. 1 The current WHO pathology classification scheme consists of clear cell carcinoma (~80%), papillary (~15%), chromophobe (~3%), collecting duct (<1%) histology and unclassifiable tumors. 2 The incidence of renal cancer has been increasing about 2% per year for the last 2 decades. 3 From an epidemiologic standpoint, this increase remains poorly understood. A number of relatively uncommon inherited syndromes are associated an increase the risk of renal cancer (e.g. Von Hippel-Lindau syndrome). 4 Environmental factors, such as hypertension, 5 smoking, 5 metal smelting, 6 and solvent exposures,7,8 and acquired or congenital cystic renal disease 9 each cause a modest increase in relative risk. It has also been suggested that obesity 5 and prosthetic joint replacements 10 produce a small increment in the risk of developing RC.
An increasing percentage of RC is incidentally discovered on CT, MRI scans and ultrasound examinations performed for unrelated reasons. 11 Despite this, approximately 25%-33% of patients present with metastatic disease at the time of initial diagnosis. In addition, 30%-40% of surgically resected patients will eventually relapse with metastatic disease. Treatment of metastatic RC remains a therapeutic challenge. With an increasing understanding of molecular biology of RC, new “targeted” therapies such as vascular endothelial growth factor receptor (VEGFR) tyrosine kinase inhibitors (sorafenib and sunitinib) have been developed in the past decade. These have markedly altered the treatment options for advanced RC.
Temsirolimus (Torisel, Wyeth Pharmaceuticals), an inhibitor of the mammalian Target of Rapamycin (mTOR), is a useful addition to the therapeutic armamentarium for the treatment of advanced RC. Temsirolimus received approval by the US Food and Drug Administration (FDA) for the treatment of renal cancer in May of 2007. The purpose of this review is to overview the mechanism and pharmacology of this agent, as well as the data supporting the use of this agent for the treatment of metastatic RC.
Chemical Structure
Temsirolimus is a derivative of the immunosuppressive macrolide rapamycin (sirolimus). Rapamycin was first discovered as a product of the bacterium Streptomyces hygroscopicus from a soil sample from Easter Island (“Rapa Nui” in Polynesian, hence the name rapamycin). The parental drug, sirolimus has been studied as an anticancer agent. Administration of sirolimus as an oral solution or tablet is characterized by a rapid but poor oral absorption (oral availability = 14%) and poor solubility in aqueous solutions. Sirolimus distributes widely in tissues (large apparent volume of distribution) and display a prolonged terminal half-life, associated with multiple biotransformations including demethylations and hydroxylations. As a result of its complex metabolism and pharmacokinetic profile, sirolimus displays large intersubject and intrasubject variability. In addition, rapamycin is immunosuppressive, and may, therefore, predispose patients to opportunistic infection. There have been anecdotal reports of activity in renal cancer. 12 A number of chemical modifications of sirolimus have been performed to increase bioavailability for therapeutic use. Temsirolimus (Rapamycin 42-[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate]) was modified from rapamycin to decrease immunosuppressive properties and to increase solubility in aqueous solution (Fig. 1). Temsirolimus has a MW = 1030.30 and molecular formula of C56H87NO16.
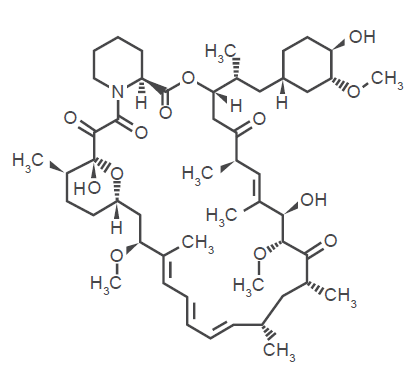
Chemical structure of temsirolimus.
Mechanism
The mTOR signaling pathway is highly conserved in yeast and mammalian cells.13–16 A simplified overview of this pathway is provided (Fig. 2). It should be noted that virtually every step in this pathway is regulated in a complex fashion by other intracellular pathways and perhaps by subcellular localization of pathway components (e.g. in association with lipid rafts in cell membrane). This complex level of control suggests a rheostat-like fine-tuning of pathway activity. In general terms, the mTOR signaling pathway links cell surface receptors (e.g. the insulin receptor and insulin-like growth factor receptor-1, IGF-1R), phosphoinositide-3-kinase (PI-3K), and the serine-threonine kinase AKT and results in downstream mTOR activation. Frequent mutations in the phosphatase and tensin homolog (PTEN) phosphatase further dysregulate the mTOR pathway, and lead to increased AKT activation. The mTOR pathway responds to growth factors (insulin, IGF-1) by recruitment of PI-3K and pAKT into ceramide-enriched lipid rafts in the cell membrane. 17 Activation of AKT results in phosphorylation of the tuberous sclerosis complex (TSC), inhibiting its GTPase activity. TSC inactivation results in accumulation of Rheb-GTP and enhancement of mTOR phosphorylation. In mammalian cells, phospho-mTOR appears to participate in two distinct multi-protein TOR Complexes (TORC).
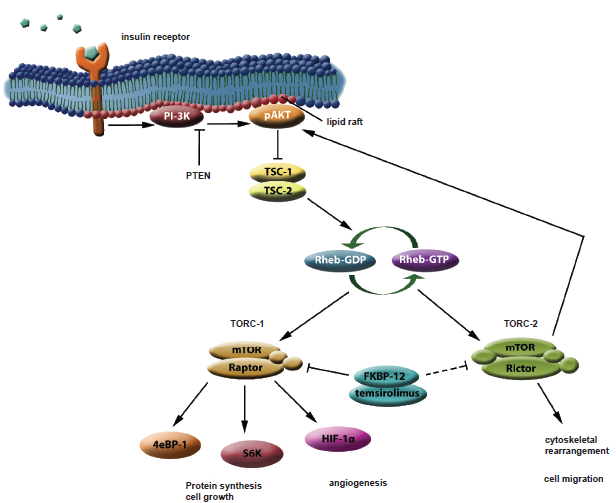
A simplified model of the TORC-1 and TORC-2 signaling pathways.
Dynamic association of mTOR with these accessory proteins results in a multitude of downstream signaling activities. When mTOR associates with a protein named Raptor, the TORC-1 complex is formed. If mTOR partners with Rictor, a distinct complex, TORC-2 is formed. TORC-1 formation is inhibited by rapamycin (and temsirolimus), while TORC-2 is generally thought to be unaffected. Cellular activity of temsirolimus requires obligate interaction with an intracellular binding protein (FKBP-12). Formation of this protein-drug complex inhibits the heterodimerization of mTOR with Raptor, resulting in downstream signaling inhibition. TORC-1 is involved in regulation of cellular energy metabolism, including ribosome biogenesis, protein translation, nutrient import, and consequently, cell growth. These effects are mediated via phosphorylation of S6 kinase (S6K) and elongation factor 4E binding protein (4eBP-1). The net effect of TORC-1 activation is to stimulate entry of cells into G1 phase of the cell cycle. 18 Indirect signaling via TORC-1 also serves to increase angiogenesis via cellular expression of hypoxia inducible factor-1α (HIF-1α). 18 In contrast, TORC-2 is involved in spatial coordination of cells, including cytoskeletal regulation and directed migration of cells. 19 It is not yet known whether the TORC-2 pathway can be directly activated by extracellular signals or receptors. It should be noted that TORC-2 acts to phosphorylate AKT at a site distinct from that phosphorylated by PI-3K, further increasing the amplitude of AKT signaling. 20 Since mTOR is a participant in both pathways, the inherent competition for this protein suggests a possible yin-yang relationship of TORC-1 and TORC-2. This may also represent potential temsirolimus resistance or escape mechanism. While temsirolimus predominantly inhibits TORC-1, combined inhibitors of both TORC-1 and TORC-2 (e.g. P529, Palomid) have recently been described. 21
Pharmacokinetics
Temsirolimus is metabolized by cytochrome P450 enzyme Cyp 3A4 to its active metabolite, sirolimus (rapamycin). The pharmacokinetics of temsirolimus has been characterized in humans.22,23 The maximal concentration of temsirolimus was detected at the end of a 30 min infusion. Maximum concentration and area under the concentration-time curve increase proportionally with dose. 24 Mean steady-state volume of distribution ranged from 127 to 385L. Whole blood clearance was nonlinear, ranging from 19 to 51 L/h (34 to 220 mg/m2). Variability predicted with flat doses appears comparable with data based on body-surface area-normalized treatment. Mean t1/2 was approximately 13 hours for temsirolimus and 60 hours for the major active metabolite, rapamycin. Exposure to rapamycin was higher than temsirolimus, with a mean area under curve ratio rapamycin/temsirolimus) of 2.5 to 3.5. 24 This indicates that exposure to the metabolite was longer than the parent drug due to its longer half-life. Drug clearance was primarily via the fecal route.
Since temsirolimus is metabolized via P450 Cyp3A4, enzyme-inhibiting drugs such as ketoconazole, clarithromycin, indinivir or enzyme inducers such as phenytoin, phenobarbital, rifampicin will alter temsirolimus metabolism and should be avoided during temsirolimus treatment. 25 A dose adjustment should be strongly considered if those medications must be co-administered during treatment with temsirolimus. Temsirolimus is considered Category D in pregnancy. Toxicity due to intrauterine growth inhibition is very likely, however, as fetal and intrauterine toxicity has been observed following oral administration of temsirolimus to rats and rabbits. 26 Therefore, women of childbearing age should be advised to avoid pregnancy during the course of treatment and 3 months after discontinuation of the drug. It was not clear whether temsirolimus is excreted in human breast milk. Therefore, this agent should not be administered to nursing mothers.
Safety, Tolerability and Efficacy
Safety, tolerability and efficacy of temsirolimus has been evaluated in phase I, II, and III clinical trials. In phase I testing, escalating doses of temsirolimus (7.5 to 220 mg/m2) were administered as a weekly infusion over 30 minutes to 24 patients with advanced solid tumors. 24 The starting dose was 7.5mg/m2, escalating up to the maximum tolerated dose, which was defined as the dose level at which ≥33% of the patients experienced dose-limiting toxicity (DLT). DLT was defined as ≤ grade 3 non-hematological toxicity (excluding alopecia, untreated nausea and vomiting), and/or hypertriglyceridemia (< 1,500 mg/dL) that recovered within 1 week, or grade 4 thrombocytopenia or neutropenia lasting >5 days, febrile neutropenia requiring hospitalization, or a treatment delay of >2 weeks as a result of unresolved toxicity.
This study showed that temsirolimus was well tolerated without serious adverse events. No clearly identified dose-limiting toxicity was established. The most frequent side effects were acne like maculopapular rashes, mucositis and stomatitis, which did not require dose reduction. Grade 3 thrombocytopenia, asthenia and diarrhea were noted in one patient, at the dose of 45 mg/m2. At dose level of 220 mg/m2, psychiatric disorders, such as euphoria and depression, stomatitis (grade 3) and asthenia (grade 3) were noted, leading to termination of the planned dose escalation. All drug related toxicity resolved after discontinuation of temsirolimus. Thus a fixed doses of temsirolimus (25 mg, not corrected to body surface area) was utilized in subsequent clinical trials.
To evaluate the effect of temsirolimus on tumor response, time to tumor progression, survival and adverse events, a phase II trial was conducted in 111 patients with advanced RC. 27 Patients were eligible if they had histologically confirmed advanced RC that had progressed following previous immunotherapy for advanced disease. Patients were required to have measurable disease, age ≥ 18, adequate renal function, hepatic and hematological function, a serum cholesterol ≤350 mg/dl and serum triglycerides ≤300 mg/dl. Patients also were required to have an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1 and a life expectancy of at least 12 weeks. Exclusion criteria included a history of CNS metastases, hepatic enzyme-inducing anticonvulsants, surgery or local radiotherapy within 3 weeks or chemotherapy, biologic therapy, or investigational drug use within 4 weeks of treatment start; prior malignancy, active infection, known HIV infection, use of immunosuppressive agents including systemic corticosteroids; significant cardiovascular disease or a history of life-threatening arrhythmia; or hypersensitivity to macrolide antibiotics. Women who were pregnant, nursing, or of childbearing potential (and not using an effective contraceptive method) were ineligible.
Patients were randomized to receive a flat dose of temsirolimus 25 mg, 75 mg or 250 mg i.v. weekly, until evidence of disease progression or unacceptable toxicity occurred. Toxicity was monitored and graded by the National Cancer Institute Common Toxicity Criteria 2.0 (CTC). A 25% dose-reduction was instituted for a decrease in ANC below 1000/μl, platelet counts below 80,000/μl, or grade 3 non-hematologic AE. A 50% dose reduction was mandated if there was a decrease in ANC to less than 750/μL or platelet count less than 50,000/μL, or a grade 4 nonhematological AE. A maximum of two doses reduction was allowed before patients were withdrawn from the study. Clinical evaluation was performed at baseline and at 4-week intervals during the course of therapy and tumor size was assessed at 8 weeks intervals.
Complete response (CR), partial response (PR), and stable disease (SD) were identified according to WHO guidelines. In addition, a minor response (MR) was defined as a 25%-50% decrease in the products of the two greatest perpendicular diameters of all measurable lesions.
The objective response rate (CR + PR) in this phase II trial was noted to be 7% (95% CI, 3.2 to 23.7). One patient had a CR and 7 patients had PRs. Additional 29 patients (26%) had MRs and 19 patients (17%) had the stable disease of >6 months duration. The study concluded that 51% of the patients achieved clinical benefit (objective response or stable disease).
Drug related toxicities were modest, and included maculopapular rash (76%), mucositis (70%), asthenia (50%), nausea (43%), hyperglycemia (17%), hypophosphatemia (13%), anemia (9%), and hypertriglyceridemia (6%). There was no apparent relationship between the temsirolimus dose and either toxicity or tumor response. Atkins et al. retrospectively characterized patients into Motzer “good”, “intermediate” or “poor” risk categories. The greatest benefit appeared to be derived in patients with “poor risk” criteria (KPS <80%, no prior nephrectomy, <1 year from diagnosis, anemia, hypercalcemia and elevated LDH).
Since the tumor response rate and median survival were comparable in three different doses and more dose reductions and discontinuations from treatment related AE were seen at the higher dose levels, the lowest dose (25 mg) was selected to use as a reference arm in a phase III trial in metastatic RC.
Combination therapy employing increasing doses of temsirolimus with either 6 or 9 million units of IFNα was tested in 71 patients with advanced RC by Motzer et al. 28 This study demonstrated that the maximum tolerated dose of temsirolimus 15 mg intravenously once a week in combination with IFNα 6 MU subcutaneously three times a week resulted in an acceptable safety profile. Tumor activity was seen in this combination, thereby establishing a second treatment arm for a phase III trial.
In the Phase III trial, 626 patients with previously untreated, poor-prognosis metastatic RC were randomly assigned to receive temsirolimus 25 mg weekly (as a 30 min intravenous infusion) or IFNα alone 3 million Units s.c. three times weekly (escalated up to 18 million U three times a week, based on patient tolerance) or a combination of temsirolimus 15 mg IV weekly i.v. plus 6 million U of IFNα s.c. three times weekly. 29 Eligible patients were required to have at least three of the following six predictors of short survival: a serum lactate dehydrogenase level ≥ 1.5 times the upper limit of the normal range, anemia, a corrected serum calcium level ≥ 10 mg per deciliter, a time from initial diagnosis to randomization ≥ 1 year, or a Karnofsky performance score of 60% or 70%. The study was later amended to also include patients with multi-organ metastases. Patients were stratified by age, sex and performance status. The primary end point of the study was overall survival (OS) and secondary end points were response rates and progression free survival (PFS) determined by both investigator evaluation of radiographs and an independent review panel. Treatment was continued as long as there was no disease progression, intolerable toxicity or symptomatic deterioration. Response valuation was by RECIST criteria.
This study demonstrated that median overall survival was increased to 10.9 months in temsirolimus-only group, compared to 7.3 months in the IFNα group, and 8.4 months in combination therapy group. The hazard ratio for death was 0.73; 95% confidence interval [CI], 0.58 to 0.92; P = 0.008, comparing the temsirolimus alone vs. IFNα alone groups. A low rate of objective response (8.6%, 4.8%, and 8.1%, respectively) was noted. These was felt to be due to the fact that the mTOR inhibitors are predominantly cytostatic and are less likely to induce objective tumor responses. Median time to treatment failure was 3.8 months for temsirolimus alone, 1.8 months for IFNα, and 2.5 months for the combination of temsirolimus with IFNα. In contrast, the proportion of patients with stable disease for ≥6 months was greater in the temsirolimus group (32.1%) versus IFNα (15.5% p = 0.002) or in the combination-therapy group (28.1%) (P < 0.001vs IFN, respectively). Subset analyses suggested that patients under 65, with Motzer poor risk (rather than intermediate risk) and non-clear cell tumors derived greatest benefit from temsirolimus treatment. 29
Asthenia (fatigue, weight loss) proved more common in the two groups receiving IFNα (alone or in combination with temsirolimus). 30 Significantly, only 11% of patients in the temsirolimus group developed asthenia compared to 26% and 28% in IFNα alone and combination-therapy group respectively (P < 0.001). Patients who developed dyspnea, diarrhea, nausea, or vomiting were similar in the three groups. Rashes, peripheral edema, and stomatitis were more common in patients who received temsirolimus, either alone or in combination with IFNα.
Hematological adverse events such as anemia, neutropenia, and thrombocytopenia were significantly more common in the combination-therapy group than in the IFNα group (P < 0.001) or temsirolimus group. In contrast, the metabolic side effects such as hyperglycemia, hypercholesterolemia, and hyperlipidemia were more common in the temsirolimus group reflecting effects on mTOR-regulated glucose and lipid metabolism. The adverse events of temsirolimus were tolerable and proved manageable with supportive care. Dose reductions and delays were less common in the temsirolimus alone group. Grade 3 or 4 AE were less common with temsirolimus (67%), compared to IFNα (78%) or combination therapy (87%) (P = 0.02).
Dutcher et al. performed a post-hoc analysis of the phase III trial, 31 evaluating the role of tumor histology, age and prognostic-risk group on outcome. The study found improved progression-free survival in both clear cell tumors and non-clear cell tumors. A substantial risk reduction in patients under 65 years (38%) and in patients with Motzer intermediate and poor-risk criteria (30%) was seen, compared to IFNα. The study concluded that all renal cancer patients (clear cell and non-clear cell) with intermediate and poor risk features gained benefit from temsirolimus treatment, regardless of histology or age. In particular, temsirolimus was recommended as the agent of choice for treatment of RC with non-clear cell histology.
Quality of Life
A quality of life analysis was also performed in conjunction with the phase III trial. 32 The study analyzed quality-adjusted time without symptoms or toxicity (Q-TWIST). Overall survival was divided into time with serious toxicity, time with progression and time without symptoms and toxicity (TWiST). Study questionnaires were available from 87% patients at progression and 40% at the time of grade 3 or 4 adverse event. Patients receiving temsirolimus alone had 38% greater time without symptoms or toxicity (TWIST) (TEMSR = 6.5 months vs. IFN = 4.7 months, p = 0.00048), 23% greater Q-TWIST score (TEMSR = 7.0 months vs. IFN = 5.7 months; p = 0.0015) than patients receiving IFNα alone. There was no difference between IFNα alone and in the combination arm in both TWIST and Q-TWIST.
Conclusion
During the past three years, substantial progress has been made in the treatment of advanced renal cell carcinoma. Single-agent temsirolimus has improved the overall survival, progression free survival and time to treatment failure with acceptable toxicity in patients with Motzer “poor risk” RC. Thus temsirolimus represents a preferred first-line therapy for patients with “poor risk” characteristics or aggressive non-clear cell RC. Empiric data is also accumulating concerning effectiveness of this agent as a salvage therapy following failure of one or more tyrosine kinase inhibitors (e.g. sorafenib, sunitinib).
Efforts to combine temsirolimus with other agents targeting other pathways (e.g. VEGF) are currently underway. Unlike efforts to block multiple steps in the VEGF pathway (vertical blockade), this horizontal blockade approach (e.g. in combination with bevacizumab, sorafenib or sunitinib) appears feasible with acceptable toxicity. Whether therapeutic results (e.g. progression-free survival) will be improved enough to warrant the increased toxicity of these combinations remains to be established.
Further work is necessary to better define the in vivo mechanisms of TORC-1 inhibitors, such as temsirolimus, in order to better understand resistance mechanisms. It is not yet clear whether the major mechanism is through inhibition of cellular protein synthesis and growth via the S6-kinase and 4eBP-1, or alternatively through indirect effects on angiogenesis via HIF-1α inhibition. Mechanisms of TORC-1 inhibitor resistance are as yet poorly defined. Thus, salvage strategies for patients that have progressed on first-line temsirolimus treatment are also just beginning to be evaluated (e.g. the AKT inhibitor perifosine, Keryx).
The success of temsirolimus has led to the development of other analogues of rapamycin. Everolimus is an orally bioavailable analogue. Phase I–II data suggested activity in metastatic RC, with a similar toxicity profile to other mTOR inhibitors. 33 In a recently published phase III study, 410 patients who have failed at least one kinase inhibitor were randomized in a 2:1 fashion to receive everolimus versus placebo. 34 Preliminary results at interim analysis resulted in early study closure, because significantly improved progression-free survival (4.0 months versus 1.9 months) was seen. No statistically significant survival benefit has yet been observed. The temsirolimus and everolimus studies have identified apparent class effects of the mTOR inhibitors (mucositis, hyperglycemia, hypertriglyceridemia and interstitial pneumonitis). While the latter appears rare, it is important to recognize this finding on CT scans, as interstitial pneumonitis can progress and be fatal.
Additional studies have indicated promising clinical activity of mTOR inhibitors in endometrial cancer, 35 neuroendocrine carcinomas, 36 sarcoma, 37 metastatic breast cancer, 38 and relapsed or refractory NHL.39,40 It is anticipated that the mTOR inhibitors will lead to further improvement in the survival of patients with metastatic RC and provide additional effective treatment options for a number of other cancers.
Disclosures
The authors have no conflicts of interest to disclose.