
Abstract
Oritavancin is a semisynthetic lipoglycopeptide with in vitro activity against a variety of aerobic Gram-positive pathogens (including drug-resistant forms of staphylococci, streptococci, and enterococci) and select anaerobic organisms. Available published clinical efficacy and safety studies in humans to date focus primarily in the treatment of complicated skin and skin structure infections. While oritavancin doses in these studies varied, single daily doses of 200 mg (300 mg in patients > 100 kg) for 3–7 days have demonstrated efficacy similar to comparators (such as vancomycin followed by cephalexin). The most frequent adverse events reported to date include gastrointestinal complaints, insomnia, dizziness, itching, and rash. Further safety and efficacy data are needed to better define its potential place in therapy.
Introduction
Skin and skin structure infections are a common clinical problem which occur in both the hospital and community settings and cause significant morbidity.1,2 Disease severity ranges from mild, superficial infections to life-threatening diseases (i.e. necrotizing fasciitis). 3 Uncomplicated infections are generally superficial and can be treated with surgical incision or oral antibiotics. 4 In contrast, complicated skin and skin structure infections (cSSSIs) are usually found in deeper tissue and are at times accompanied by systemic signs and symptoms of infection (i.e. fever, tachycardia, hypotension). 5 Such infections often require hospitalization, parenteral antimicrobial therapy, and surgical intervention. 6 According to data from the National Hospital Discharge Survey, such infections were the primary admission diagnosis for approximately 780,000 hospitalizations in 2006. 1
The majority of cSSSIs are caused by Gram-positive bacteria such as Staphylococcus aureus, enterococci, and B-hemolytic streptococci. 7 S. aureus is the predominant pathogen causing these infections. For example, over a 7-year period (1998–2004), 44.6% (2602/5837) of isolates from North American patients with skin and skin structure infections were due to S. aureus. 8 Particularly problematic are infections caused by methicillin-resistant S. aureus (MRSA) and vancomycin-resistant enterococci (VRE). The rate of MRSA in the aforementioned study increased from 40.5% in 2002 to 51.6% in 2004, while VRE almost doubled over the same time period (8.6% to 14.8%). 8
Prompt appropriate antibiotic therapy in patients with cSSSIs has been associated with decreased length of stay and total costs. 9 The “gold standard” for these infections has been intravenous vancomycin, especially when MRSA is considered a likely pathogen. However, recent concerns regarding the potential for increased clinical failure of vancomycin therapy in the setting of rising minimum inhibitory concentrations (MICs) have stimulated the need to examine alternative therapies.10–13 Alternative agents currently FDA-approved for cSSSIs include linezolid, daptomycin, and tigecycline. However, these agents are not without their limitations, including need for parenteral therapy (daptomycin, tigecycline), adverse effects, and increased drug acquisition costs. Therefore, there is a continued need to develop safe, efficacious and cost-effective treatment options for cSSSIs.
Oritavancin (LY333328 diphosphate; The Medicines Company®) a semisynthetic lipoglycopeptide, is an intravenous antibiotic developed to treat serious infections caused by Gram-positive pathogens, including vancomycin-resistant organisms. To date, the safety and efficacy of oritavancin for the treatment of cSSSIs have been evaluated in Phase II and III studies.14,15 The purpose of this article is to review the pharmacologic and microbiologic properties of oritavancin and summarize available clinical efficacy and safety data for the treatment of cSSSIs.
Pharmacology
Oritavancin is a semisynthetic lipoglycopeptide antibiotic structurally similar to vancomycin. 16 It is derived from the naturally occurring compound, chloroeremomycin. 16 Oritavancin appears to have multiple mechanisms of action.16–18 Similar to vancomycin, oritavancin inhibits peptidoglycan synthesis by binding to the D-Ala-D-Ala terminal end of peptidoglycan precursors, resulting in the inhibition of cell wall synthesis. 19 However, recent solid-state nuclear magnetic resonance experiments propose that oritavancin significantly inhibits transpeptidation in S. aureus and E. faecium, while vancomycin primarily inhibits transglycosylation.18,19 In addition to the primary D-Ala-D-Ala binding interaction, in vitro data have suggested that oritavancin also binds to secondary cell wall binding sites.18,19 These proposed secondary binding sites are the pentaglycyl bridging segment in S. aureus and the triangulation of a D-Asx bridge, an L-Lys, and a penultimate D-ala4 formed on a pentapeptide stem in E. faecium. This unique, dual mode-of-action may contribute to the enhanced bactericidal activity against vancomycin-resistant S. aureus and enterococci. 18 In addition to its primary mechanism of action, oritavancin also disrupts membrane permeability17,20 and inhibits RNA synthesis. 16
In vitro data demonstrate that oritavancin possesses a sustained, concentration-dependant post-antibiotic effect (PAE). 21 A PAE of 1.5 to 2 hours was reported in a vancomycin intermediate S. aureus (VISA) strain, while PAEs of 3 to 4.5 hours were reported in strains of VRE and vancomycin-resistant S. aureus (VRSA). Various in vivo and in vitro studies have also described other pharmacodynamic properties of oritavancin. Similar to daptomycin, oritavancin exhibits concentration-dependent, bactericidal killing. 22 Based on data from a neutropenic murine thigh-infection model, the ratio of the area under the 24 hr time-concentration curve (AUC0–24) to the minimum inhibitory concentration (MIC) of (AUC0–24:MIC ratio) and AUC0–24 are the pharmacodynamic parameters which best correlate with efficacy against S. aureus, while AUC0–24 best correlates with efficacy against Streptococcus pyogenes23,24 Using the 48hr timepoint data, mean AUC0–24: MIC ratios associated with 1-, 2-, and 3-log10 CFU reductions of S. aureus from baseline were 87.9, 99, 111, and 131, respectively 23 The median AUC0–24 values associated with the same endpoints with Streptococcus pyogenes at 48 hours were 3.87, 5.16, 6.93, and 9.78, respectively. 24
Pharmacokinetics
Single- and multiple-dose studies in humans have been performed to describe the pharmacokinetic properties of oritavancin.25,26 Oritavancin displays linear pharmacokinetics following both weight-based (0.02 to 10 mg/kg) and fixed-dose regimens (100 to 800 mg).25,26 The median (range) Cmax and AUC0–t reported from 4 patients receiving 0.5 mg/kg was 6.5 (4.7 to 7.6) mcg/mL and 68.3 (46.7 to 77.4) mcg x hr/L, respectively, 26 while terminal half-life is approximately 320 hours. 27 Oritavancin is highly protein-bound (~82%). 28 Animal studies evaluating tissue distribution of oritavancin have shown that approximately 59 to 64%, 2.7%, 1.8%, and 1.7% of the administered dose are found in the liver, kidney, spleen, and lung, respectively 29 In a blister fluid model, mean drug concentrations exceed the oritavancin MIC at which 90% of strains are inhibited (MIC90) for S. aureus (2 mcg/mL) by approximately 2- to 5.5-fold at 12 hours and 1.5- to 3-fold at 24 hours following administration of 200 mg of oritavancin once a day for 3 days and 800 mg as a single dose. 25 The study's investigators speculated that such concentrations in the blister fluid were likely to be adequate to treat cSSSIs.
To date, there is no evidence that oritavancin undergoes metabolism. 29 Elimination of oritavancin occurs through both the renal and fecal routes. A dose-escalating study reported <5% and 1% of administered drug were recovered in the urine and feces (respectively) 7 days after a single dose. 26
Spectrum of Activity
There are currently no published breakpoint criteria from the Clinical and Laboratory Standards Institute (CLSI) to determine susceptibility or resistance for oritavancin. To determine the activity of oritavancin, there are several laboratory conditions that first must be considered. The CLSI currently recommends to include polysorbate 80 for broth microdilution for susceptibility testing of oritavancin because of significant binding that occurs with oritavancin to plastic surfaces.30–33 A recent study demonstrated that when polysorbate 80 was used in the in vitro testing of oritavancin to Enterococci spp. and Staphylocci spp., a reduction in MIC90s of 16–32 times was noted. 30 Therefore, previous studies not utilizing polysorbate 80 may have underestimated the in vitro efficacy of oritavancin against these isolates.30–33
In addition to issues regarding polysorbate 80, a recent study evaluated other conditions that may affect susceptibility in vitro 34 This included incubation time, frozen panel storage, pH and inoculum size. Incubation in carbon dioxide as well as the presence of calcium ions were also evaluated. E. faecalis, S. aureus, S. pneumoniae were all tested by broth microdilution in the presence of polysorbate 80. Inoculation time (up to 48 hours), frozen panel storage time (up to 6 months), carbon dioxide incubation, and calcium concentration (50 mcg/mL) were all noted to have equivalent or up to 1 twofold dilution MICs (as compared with the CLSI parameters) for all three isolates. When inoculum size was increased from standard 5 x 10 5 CFU/mL to 5 x 10 7 CFU/mL, 2 twofold dilutions increase of MICs were seen for E. faecalis and 3 twofold dilutions for S. aureus. For S. pneumoniae, oritavancin MICs were not affected by increasing inoculum. No growth effect was observed for either S. pneumoniae or E. faecalis when altering the medium pH to either 5.2 or 9.2. Therefore, MICs could not be established. In contrast, such changes increased MICs against S.aureus by up to 2 twofold dilutions. 34
Because oritavancin is highly protein-bound (>80%), albumin is another factor that must be considered. 35 When tested in bovine serum albumin, oritavancin showed reduced activity (99.9% verses 5% killing of S. aureus with oritavancin for control and bovine serum albumin media, respectively) as well as a shorter post-antibiotic effect (65 minutes vs 5 minutes, respectively). 35 Further study is warranted to determine if this finding is clinically relevant.
Like the other glycopeptides, oritavancin is active in vitro against most Gram-positive aerobic and anaerobic organisms, while lacking activity against Gram-negative organisms (see Table 1).36,37 Activity against Gram-positive organisms includes drug-susceptible and -resistant strains of S. aureus (oxacillin-susceptible and -resistant isolates) and Streptococcus pneumoniae (including penicillin-susceptible, intermediate and resistant organisms). It also appears to be active against Listeria spp. with an MIC90 = 0.06 mcg/mL (range ≤0.03–0.125 mcg/mL)36,38 In addition, oritavancin also shows favorable activity against both vancomycin-susceptible and -resistant strains of enterococci, including both E. faecalis and E. faecium. For vanA (n = 33) strains of VRE, the MIC range was reported to be 0.06–1 mcg/mL (MIC90 = 1 mcg/mL). 36 For vanB (n = 5) and vanC (n = 20) strains, the MIC ranges were 0.06–0.5 mcg/mL (MIC90 = not reported) and ≤0.03–1 mcg/mL (MIC90 = 0.5 mcg/mL), respectively. 36
In vitro activity of oritavancin.
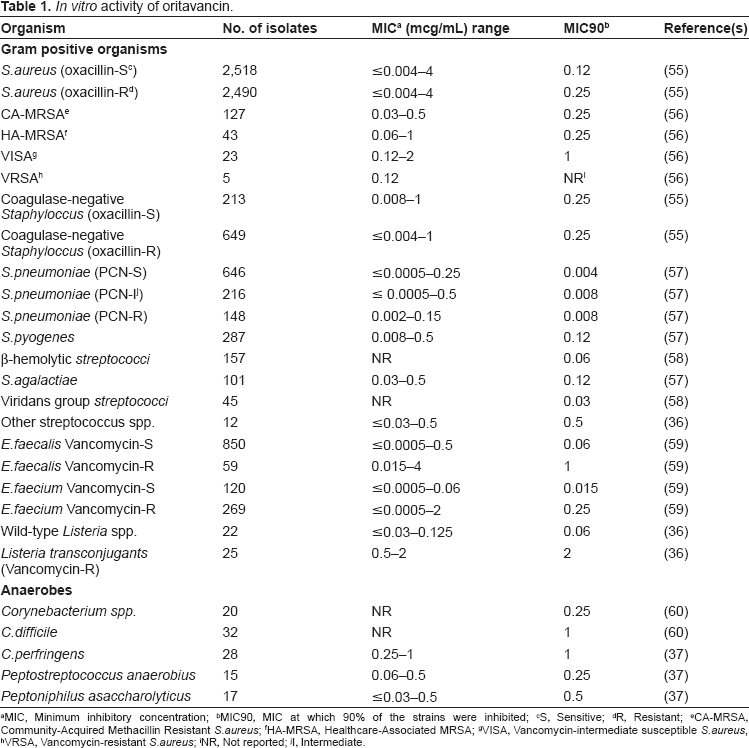
MIC, Minimum inhibitory concentration
MIC90, MIC at which 90% of the strains were inhibited
S, Sensitive
R, Resistant
CA-MRSA, Community-Acquired Methacillin Resistant S.aureus
HA-MRSA, Healthcare-Associated MRSA
VISA, Vancomycin-intermediate susceptible S.aureus
VRSA, Vancomycin-resistant S.aureus
NR, Not reported
I, Intermediate.
In addition to Gram-positive aerobic activity, oritavancin displays potent anaerobic activity as well. When tested against Clostridium perfringens (n = 28) and Peptrostreptococcus anaerobius (n = 15), the MIC range and MIC90 for oritavancin was 0.25–1 and 1 mcg/mL, and 0.06–0.5 and 0.25 mcg/mL, respectively. 37 Against C. difficile, an MIC range of 0.125–2 mcg/mL and MIC90 of 1 mcg/mL was reported. 36 Oritavancin (unlike vancomycin) also displayed activity against the C. difficile spore forms of the organism. 39
Animal Models of Infection
Skin and skin structure infections
To date, there are no published studies evaluating oritavancin in the treatment of skin and skin structure infections in the animal model. However, there are other types of infections describing the efficacy of oritavancin that are described below.
Other infections
Oritavancin has been studied in animal models of endocarditis, meningitis, catheter-related infections and inhalation anthrax.40–44 In the endocarditis model, rabbits were inoculated with vancomycin-susceptible and -resistant (VanA and VanB) E. faecalis 41 The oritavancin MICs for all three organisms were 2 mcg/mL. Oritivancin (20 mg/kg IV once daily for five days) was studied both alone and in combination with gentamicin (3 mg/kg intramuscular twice daily for five days). While oritavancin demonstrated efficacy against the vancomycin-susceptible strain, it was not active as monotherapy for the resistant strains. In fact, resistance emerged in the models with VanA and VanB strains in this study. 41 However, oritavancin in combination with gentamicin was active against all three strains.
There are currently two published studies evaluating oritavancin in the rabbit model for the treatment of meningitis.42,43 These studies focused on both penicillin-susceptible S. pneumoniae as well as the cephalosporin-resistant S. pneumoniae42,43 The results of this study showed that single doses of oritavancin (2.5 mg/kg and 10 mg/kg) were effective in reducing the bacterial burden in the cerebrospinal fluid similar to ceftriaxone (control). Oritavancin 2.5 mg/kg and 10 mg/kg reduced the log colony-forming units (CFUs) by 0.26 ± 0.22/ml/h and 0.29 ± 0.21/ml/h, respectively. The reduction for ceftriaxone (10 mg/kg/h) was 0.34 ± 0.15/ml/hr. In the resistant-pneumococcal meningitis study, 42 the rabbits were given dexamethasone 0.25 mg IV x 1 or saline followed ten minutes later by one of the following: oritavancin 10 mg/kg/day or oritavancin 10 mg/kg/day plus ceftriaxone 100 mg/kg/day. The control was given ceftriaxone. The results demonstrated a marked reduction in CFUs at both six and 24 hours for the oritavancin alone groups as well as the combination oritavancin plus ceftriaxone, although synergy was not demonstrated. 42
Oritavancin has also been studied in the rat model for catheter-associated infections. 40 Rats inoculated with vancomycin-resistant (VanA) E. faecium via a 15 minute dwell time for the central venous catheters were given either a single dose of oritavancin 20 mg/kg (n = 8) or saline (n = 8). The rats were then sacrificed eight days later. The results revealed that the infection rate (defined as VRE bacteremia or isolation of the organism from the catheter or organs) was much higher for the saline-treated rats compared with the oritavancin-treated rats when evaluating the catheter (87.5% vs. 12.5%, respectively), peripheral blood or heart (75% vs. 0%, respectively), lung or kidney (100% vs. 0%, respectively) and the liver (87.5% vs. 0%, respectively).
The use of oritavancin has been evaluated in the treatment of inhalation anthrax in the murine model. 44 This study evaluated both pre- and post-exposure prophylaxis. For pre-exposure prophylaxis, mice were given a single dose of oritavancin 50 mg/kg IV 24 hours-28 days before exposure. Ciprofloxacin was used as a comparator in all three groups. Effective protection in the oritavancin group (the 24 hour and 7 day prophylaxis) was demonstrated, with 90% survival rate at 33 days post challenge. The 14-day prophylaxis also demonstrated effectiveness, with 100% survival at 22 days post challenge. In contrast, all mice in the ciprofloxacin pre-exposure prophylaxis group died. In the post-exposure group, marked protection was demonstrated for oritavancin every 48 hours x 14 days with the 1 mg/kg (50% survival rate) and 3 mg/kg (100% survival rate) groups. The 15 mg/kg and 50 mg/kg single-dose groups had survival rates of 70 and 100%, respectively. The ciprofloxacin mice had similar outcomes. In the post-exposure treatment group, symptoms of anthrax were observed as early as 36 hours post-exposure. Treatment with oritavancin revealed a survival rate of 90% and 50% in the 36-hour and 48-hour post-challenge treatment groups, respectively. Similarly, ciprofloxacin's survival rate was 70% and 80% in the 36-hour and 48-hour post-challenge treatment groups, respectively. 44
Clinical Studies
Complicated skin and skin structure infections
A phase II, randomized, double-blinded, parallel, active-comparator study (SIMPLIFI- Single or Infrequent Doses for the Treatment of cSSSI) of 302 cSSSIs patients from five countries (Australia, India, Romania, Ukraine, and United States) and forty-three centers was conducted. 15 Patients ≥18 years of age with a cSSSI suspected or confirmed to be caused by a Gram-positive organism and a body mass index between ≥17 kg/m2 and ≤40 kg/m2 were included. Patients were stratified based on their disease category (wound infection, major abscess, or cellulitis) and randomized in a 1:1:1 ratio to receive either oritavancin daily dose (200 mg daily for 3–7 days determined by the investigator based on clinical criteria), oritavancin single dose (1200 mg on day 1) or oritavancin infrequent dose (800 mg on day 1 with an optional dose of 400 mg on day 5–-at the discretion of the investigators). A majority of patients were Caucasian (63.2%) and male (65.9%) with a mean age of 45 years. Baseline characteristics and demographics were comparable between all three groups. The most commonly isolated organism was S. aureus (87.6%), of which 49.3% were MRSA. Cure rates in the clinically-evaluable population (N = 228) at the test-of-cure visit was 72.4% (55/76) for oritavancin daily dose, 81.5% (66/81) [1200–200 mg,90% CI: -2.5,18.2] for oritavancin single dose, and 77.5% (55/71) [800–200 mg,90% CI: -6.8,15.4] for the oritavancin infrequent dose. Clinical cure rates in the intent-to-treat population (N = 300) for daily, single, and infrequent dose oritavancin was 72.4% (63/87), 81.8% (72/88) [1200–200 mg,90% CI: -1.7,17.8], and 78.2% (68/87) [800–200 mg,90% CI: -5.8,14.6], respectively. Patients with MRSA (N = 83) in the clinically-evaluable population had cure rates of 78.3%, 73% [1200 mg–200 mg,90% CI: -25.1,12.9], and 87% [800–200 mg,90% CI: -6.9,25.3], respectively. Patients in all of the groups tolerated the drug well with similar rates of adverse effects, which included injection site phlebitis and histamine-like infusion reaction. However, the frequency of adverse events was not reported.
Oritavancin was also evaluated for the treatment of cSSSIs in two Phase III, randomized, double-blinded, multi-centered, active comparator studies. Results were pooled and presented in a single abstract (ARRD and ARRI studies). 14 Patients either received oritavancin intravenously daily for 3–7 days followed by oral placebo for remaining duration of the study (for a total of 10–14 days) or vancomycin 15 mg/kg intravenously every 12 hours for at least 3 days to 7 days (adjusted for renal function) followed by oral cephalexin 500–1000 mg every 12 hours for a total of 10–14 days. Patients were switched to orals once they met the protocol criteria for the switch. For patients with MRSA or Enterococcus spp at baseline, oritavancin for 7 days or vancomycin for 10–14 days was utilized. Patients in ARRD study (N = 531) were randomized in a 1:1:1 fashion to receive either oritavancin 1.5 mg/kg/day, 3.0 mg/kg/day (maximum 400 mg), or comparator drug. Patients in the AARI study (N = 1267) were randomized 2:1 to receive 200 mg/day of oritavancin (300 mg if patient > 110 kg) or comparator. 14 Patients were also further stratified based on the type of cSSSIs (i.e. wound infection, major abscess, or cellulitis). Data from both studies were pooled and two oritavancin groups (all patients receiving any oritavancin dose [Oritavancin-ALL] and those receiving a total daily dose of oritavancin between 180 mg to 330 mg [Oritavancin-DR]) were analyzed. Baseline demographics were similar between groups. Patients received a mean of 5.2 (+/-1.55) days of oritavancin therapy and 6.1 (+/-3.27) days of vancomycin. In terms of the total number of days of therapy, patients in the oritavancin arm received a mean of 5.2 (+/-1.55) days of therapy compared to 11.3 (+/-3.38) days in the comparator arm (vancomycin +/- cephalexin). The primary endpoint of clinical cure rates at the test-of-cure (TOC) in the clinically evaluable (CE) population was 76.9% (722/939), 77.7% (597/768) [Oritavancin 180–330 mg-Vancomycin,95% CI: -2.9,6.9], 75.8% (347/458) for the Oritavancin-ALL, Oritavancin-DR, and vancomycin arms, respectively. Microbiological success rates in the microbiologic evaluable (ME) population was 78.2% (448/615) for the Oritavancin-ALL arm, 74% (378/511) in the Oritavancin-DR arm [Oritavancin 180–330 mg-Vancomycin,95% CI: -4.7,7.7], and 72.5% (232/320) for the vancomycin arm. Microbiologic success in patients with MRSA were 63.2% (72/114), 63% (63/100), and 67.3% (33/49) for Oritavancin-ALL, Oritavancin-DR, and vancomycin arms, respectively. Based on the pooled analysis, the authors concluded that oritavancin 200 mg (300 mg for patients > 110 kg) for 3 to 7 days was noninferior to vancomycin/cephalexin every 12 hours for 10 to 14 days in the treatment of cSSSIs. 14
Other infections
The safety and efficacy of oritavancin in the treatment of adult patients with Gram-positive bacteremia has been evaluated in two phase II studies.45,46 An open-labeled, non-controlled trial was conducted in patients with Gram-positive bacteremia (N = 27) who received one of the following regimens for a total of 7–10 days: 3 mg/kg loading dose followed by 2 mg/kg maintenance dose (N = 5), 4 mg/kg loading dose followed by 3 mg/kg maintenance dose (N = 5), or 5 mg/kg loading dose followed by 4 mg/kg maintenance dose (N = 17). 45 Favorable bacteriologic response at the first follow-up visit five days after the end of therapy was observed in nine out of the ten evaluable patients in the 5/4 mg/kg group. No results were reported for the other treatment groups. No serious adverse effects occurred in the 5/4 mg/kg group. 45
A phase II, open-labeled, randomized, noninferiority study evaluated the efficacy and safety of oritavancin in the treatment of S. aureus bacteremia. 46 Patients (N = 125) were randomized to receive either the comparator drug (vancomycin 15 mg/kg IV q12h or a B-lactam agent if the organism was susceptible) or oritavancin (5, 6.5, 8 or 10 mg/kg IV once a day) for 10–14 days. Oritavancin was as effective as the comparator arm in the evaluable population (N = 84). Composite success (clinical cure and bacteriologic eradication) was observed in 83% (5/6), 71% (5/7), 67% (16/24), 80% (16/20) for the 5, 6.5, 8 and 10 mg/kg arms of oritavancin, respectively. The comparator arm had a composite success rate of 70% (19/27). Clinical cure rates for the oritavancin arms (5, 6.5, 8 and 10 mg/kg arms, respectively) were 83% (5/6), 71% (5/7), 71% (17/24), 80% (16/20) compared to the comparator arm of 74% (20/27). The bacteriologic eradication rates were 83% (5/6), 86% (6/7), 79% (19/24/), 85% (17/20) for the oritavancin arms (5, 6.5, 8 and 10 mg/kg arms, respectively) and 78% (21/27) for the comparator arm. Rates of deaths were similar in all treatment groups. The most common adverse effects were diarrhea (17% vs. 19%), phlebitis at the injection site (19% vs. 14%) and pyrexia (19% versus 14%) in the oritavancin and comparator groups, respectively. 46
Safety/Tolerability
The safety and tolerability of oritavancin has been studied in both healthy human subjects and patients with cSSSIs.15,26,47,48 In the first clinical study evaluating cSSSIs, 302 patients were randomized to receive oritavancin 200 mg per day for 3–7 days, a single dose of oritavancin 1200 mg on day one or a spaced dosing of oritavancin 800 mg on day one with the option of having a repeat 400 mg dose on day 5. 15 The safety profile was similar among all three groups, with more than 1 adverse event occurring in 56%, 56% and 61%, respectively. The number of patients that had to discontinue study medication was 3%, 3% and 1%, respectively. The only side effect noted was mild-moderate injection site phlebitis which occurred in similar incidences (exact incidence not reported) in all three groups. 15
Two randomized, double-blind, phase III trials examined the safety of oritavancin. 47 A total of 1,173 patients were randomized to oritavancin (dosed at 1.5 mg/kg or 3 mg/kg in one study and 200 mg per day for patients ≤ 110 kg or 300 mg per day for patients > 110 kg in the other study) or vancomycin 15 mg/kg every twelve hours (adjusted for renal dysfunction) followed by cephalexin for the treatment of cSSSIs. 47 A significantly lower incidence of treatment-related adverse effects (53%) was observe in the oritavancin group compared with 62% in the vancomycin group (p < 0.001). The most common side effects reported in this study (≥2%) included gastrointestinal complaints (nausea, vomiting, abdominal pain, diarrhea or constipation), headache, central nervous system disturbances (insomnia or dizziness), itching, rash and elevated blood pressures. Hypokalemia (≥2%) was also reported. Elevations in serum creatinine (3.7% oritavancin vs. 4.2% vancomycin) and liver function tests (defined as >3–5 x upper limit of normal) (0.4% AST, 0.8% ALT oritavancin vs. 1.1% AST, 1.5% ALT vancomycin) were similar between the two groups. 47 However, no difference as noted in the incidence of serious adverse events (9% vs. 11%, respectively), deaths (1.6% vs. 2.0%, respectively) and study-drug discontinuations due to adverse events (35% vs. 34%, respectively). Histamine-like infusion reactions (described as flushing or redness of the head, neck and upper torso and extremities, itching, rash and hypotension) occurred with a significantly lower incidence in the oritavancin group compared with the vancomycin group (3.1% vs. 11% respectively, p < 0.001).47,48
The safety of oritavancin was studied among 11 healthy subjects receiving oritavancin 0.02–0.5 mg/kg over 1 hr in eight increasing increments on eight separate days. 26 The observation time was 6 days after the last dose with outpatient follow-up for an additional 8 weeks. In addition to physical examination, laboratory studies, electrocardiogram and audiometric evaluations were performed for those receiving the 0.5 mg/kg dose. Of 10 that completed the study, most (n = 58 reactions) reported mild-moderate adverse reactions with no deaths or severe events. Headache (12 episodes) followed by rhinitis (5 episodes) and dry skin (5 episodes) were the most common reactions. Others included CNS disturbances (abnormal dreams, insomnia, anxiety) and gastrointestinal effects (diarrhea and flatulence). Five of eleven patients receiving oritavancin demonstrated transient and asymptomatic increases in AST and ALT. No subjective reports of hearing loss were reported, and audiometric studies showed no changes when comparing pre and post studies among the 3 of the 4 patients tested. The fourth patient experienced a unilateral increase in hearing at a select frequency after the study. EKG results were not reported. 26
QT interval changes from oritavancin were assessed in 240 healthy subjects. 49 Single doses of oritavancin (200 mg IV and 800 mg IV) were compared with moxifloxacin (400 mg PO x 1) and placebo. When comparing oritavancin 800 mg and placebo, no clinically significant effect on QTc for oritavancin was noted throughout the study. 49
Dosing and Administration
Various dosing regimens have been utilized in clinical studies evaluating oritavancin for the treatment of cSSSIs, including 200 mg IV daily, a single dose of 1200 mg IV, 1.5 mg/kg IV daily, and 3 mg/kg IV daily.14,15 The proposed dosing regimen submitted to the FDA for the treatment of cSSSIs is 200 mg daily for 3–7 days. 27 Data from a pharmacokinetic model proposed that obese patients (≥110 kg) receiving 200 mg daily would be expected to have lower AUC0–24 values lower than subjects weighing <110 kg due to high clearances. 29 Therefore, doses of 300 mg for patients weighing ≥110 kg may be warranted. Based on the limited available data, dose adjustments of oritavancin are not necessary for the elderly, patients with mild to moderate hepatic insufficiency (Child Pugh score 5–10), or patients with renal dysfunction.29,50,51 In addition, it appears that oritavancin is not significantly removed by hemodialysis. 52 However, caution should be used in these special patient populations until additional data is obtained.
Current Status
In February 2008, Targanta Therapeutics Corporation (acquired by The Medicines Company® in February 2009) submitted a New Drug Application with the US Food and Drug Administration (FDA) seeking approval of oritavancin for the treatment of cSSSIs. In November 2008, the FDA Advisory Committee voted against the approval of oritavancin, citing the need for an additional well-controlled study demonstrating safety and efficacy. 53 Most recently, The Medicines Company announced in August 2009 the withdrawal of the European marketing authorization application for oritavancin after the European Medicines Agency informed the company that another study would be required before approval could be considered, similar to the FDA's recommendation. 54
Potential Place in Therapy/Conclusion
With its broad-spectrum of activity against Gram-positive pathogens including multi-drug resistant organisms such as MRSA, VRSA, VRE, and PCN-resistant S. pneumoniae, oritavancin will likely be an alternative therapy to those drugs already available targeting such pathogens. In addition, oritavancin's dual mechanism of action and rapid bactericidal activity may assist in decreasing or delaying the emergence of resistance. Furthermore, oritavancin has a long half-life and a post-antibiotic effect which facilitates once daily administration and (potentially) a shorter duration of therapy in comparison to currently-available drugs. Oritavancin's favorable safety profile, lack of need for serum concentration monitoring, and lack of dosing alterations in patients with hepatic or renal insufficiency are all desirable characteristics. Clinical studies to date have demonstrated promising results for the use of oritavancin in the treatment of cSSSIs (and possibly bacteremia).
Disclosures
Drs. Townsend, Wilson and Pound–-None; Dr. Drew Research for Cubist, Schering-Plough; Consultant for Theravance, Schering-Plough, Astellas; Speaker honorarium for Wyeth, Merck, Schering-Plough, Ortho-McNeil; Development Team–-CustomID.