
Abstract
Diabetes Mellitus is a chronic condition prevalent worldwide. Type 2 diabetes is the most common form of diabetes, comprising 90% to 95% of all cases. Over the last few decades, the importance of glycemic control and its impact on prevention of diabetes-related complications has been documented in multiple clinical trials. As most patients with type 2 diabetes will require pharmacologic intervention to achieve and maintain appropriate glycemic control, new medications targeting different aspects of the pathophysiology of type 2 diabetes have been a significant focus of research and development. During the last decade, multiple new medications for diabetes management have become available: these medications have novel mechanisms of action, differences in effectiveness, and varying side effect profiles which will be reviewed in this article. Some of these newer medications, such as the GLP-1 analogues and DPP-4 inhibitors, have become widely accepted as therapeutic options for the management of type 2 diabetes. Additional classes of glucose-lowering medications are expected to become available in the near future. This manuscript will summarize available data regarding these newer and prospective medications for the management of type 2 diabetes.
Introduction
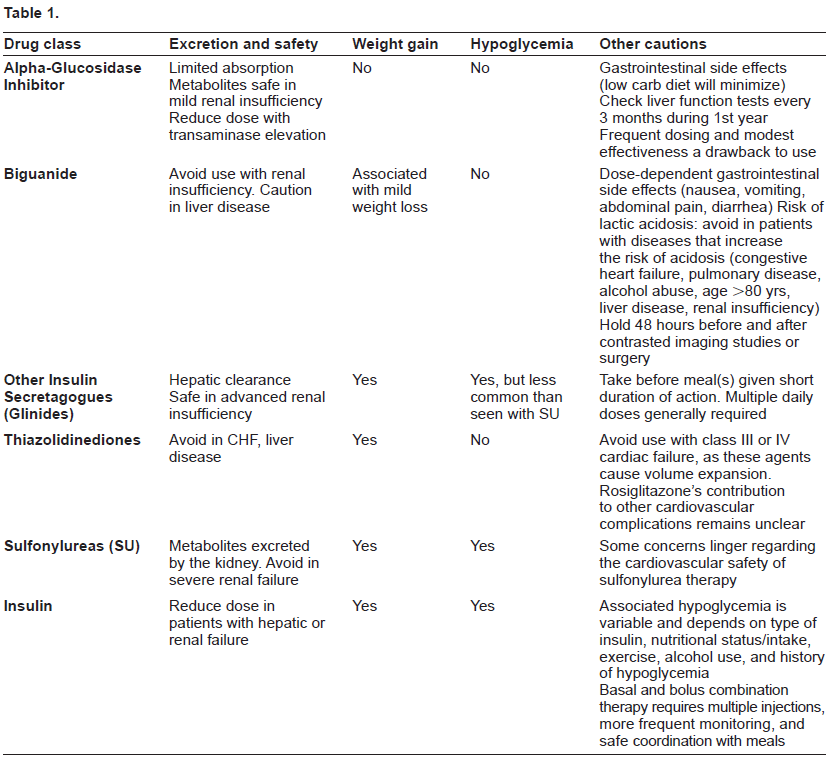
Diabetes mellitus is a chronic condition prevalent worldwide. It is estimated that more than 180 million individuals have diabetes, with this number expected to increase to 366 million by the year 2030. 1 Type 2 diabetes is characterized by both disorders of insulin activity as well as inadequate insulin production by the pancreatic beta cells, 2 and is the most common form of diabetes, comprising 90% to 95% of all diabetes cases. 3 The chronic hyperglycemia of diabetes is associated with long term damage to various organs, including the eyes, kidneys, nervous system, heart, and vasculature. In addition, it is among the leading causes of blindness and renal failure worldwide. 45 Over the last few decades, studies have shown that better glycemic control, as evidenced by lower Hemoglobin A1c (HbA1c) levels, results in significantly lower rates of microvascular complications. In the United Kingdom Prospective Diabetes Study (UKPDS), every 1% drop in hemoglobin HbA1c corresponded to a 37% reduction in microvascular complications and a 21% reduction in any diabetes-related endpoint. 6 Consequently, current recommendations focus on maintaining glycemic levels as close to the non-diabetic range as is feasible. It is clear that in order to achieve this goal, the vast majority of individuals affected by type 2 diabetes will require pharmacologic intervention, and many will require multiple agents. Therefore, new medications continue to be a primary focus of research and development. In this review, we will discuss pharmacologic agents available for the management of type 2 diabetes, but will focus primarily upon those most recently developed or in development. A brief review of key points relating to traditionally used glucose-lowering medications is available in Table 1. The remainder of this article will describe newer agents which enhance the function of the incretin system, are analogues of human hormones, and/or utilize novel mechanisms in elucidating glucose control.
Incretin-Based Therapies
Incretin hormones and the incretin effect
In 1902, Bayliss and Starling published a pivotal manuscript, “The mechanism of pancreatic secretion,” that described the behavior of the hormones now called incretins. 7 The word “incretin” itself was introduced in 1932, and, in 1964, Elrick and McIntyre separately and simultaneously described the “incretin effect.” The term describes the more robust increase in insulin response that is seen following oral glucose administration as compared to intravenous glucose administration. The effect is maintained even in the case of higher blood glucose levels during the intravenous infusion.8,9 These events led to the discovery of the major incretin hormones glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide (GLP-1). These hormones are released from the gut shortly after food ingestion, and have actions that are largely responsible for the described incretin effect. Importantly, the incretin effect appears to be blunted in individuals with type 2 diabetes. 10
GLP-1 is the most potent known incretin, with a level that begins to increase almost immediately after food intake. It binds to pancreatic receptors, resulting in release of insulin from the beta cells and suppression of glucagon release from the alpha cells. In addition, GLP-1 slows gastric emptying and promotes satiety at the level of the central nervous system. GLP-1 has also been found to stimulate growth and survival of beta cells in animal models and is thought to stimulate proliferation and differentiation of new beta cells. 11 Interestingly, GIP has a similar effect on postprandial insulin release but may have an opposite, stimulatory effect on glucagon release. 12 The effects of both GIP and GLP-1 are glucose-dependent: their glucose-lowering activity ceases when glucose levels fall below 65 mg/dl. 13 The half lives of GLP-1 and GIP are only a few minutes long, as they are rapidly degraded to inactive metabolites by dipeptidyl peptidase-4 (DPP-4), an enzyme widely expressed throughout the body. 13
Individuals with type 2 diabetes are GLP-1 deficient; however, infusion of GLP-1 to individuals with this condition has been shown to lower both postprandial and fasting blood glucose levels.13,14 Conversely, there appear to be relatively normal levels of GIP in persons with type 2 diabetes, but their physiologic response to GIP is diminished. 15 Classes of drugs currently available which primarily utilize the incretin pathway to improve glycemic control include the GLP-1 analogues and DPP-4 inhibitors.
GLP-1 Analogues
Exenatide
Mechanism of action
The first commercially available GLP-1 analogue is exenatide, a synthetic version of a peptide isolated from the saliva of the Gila monster. 16 This substance, exendin-4, has actions similar to but is more resistant to degradation by DPP-4 than is endogenous GLP-1. This structural change results in an extended halflife averaging 2.4 hours. 17 The total duration of action following subcutaneous injection has been reported to be five to seven hours in humans, 18 but may last up to 10 hours after each injection. 17 The longer half-life and decreased degradation allows for exenatide to reach reported concentrations five to ten times greater than physiological GLP-1 levels in patients with type 2 diabetes. 18 The currently available formulation of exenatide is administered as a twice daily subcutaneous injection, given up to 60 minutes prior to a meal; however, a once weekly formulation is also in late stages of development.18,19 The starting dose of exenatide is 5 μg administered twice daily, titrated up after 1 month to 10 μg twice daily based on tolerability and glycemic control. 17
Efficacy in clinical studies
Exenatide has been investigated as monotherapy and as adjunctive therapy with metformin alone, metformin plus sulfonylurea, thiazolidinedione (TZD) alone or with metformin, and insulin. 16 In monotherapy trials, exenatide 10 μg twice daily, compared to placebo, resulted in placebo-subtracted HbA1c reductions of 0.6% to 1.0%.20,21
In 2004, Buse et al compared 10 μg twice daily vs. 5 μg twice daily vs. placebo in 377 patients with mean baseline HbA1c of 8.6% on maximal sulfonylurea therapy. HbA1c improvements were dose-dependent, with placebo-subtracted HbA1c reductions of 0.98% in the high dose group and 0.58% in the low dose exenatide group. HbA1c reductions were greater in patients with baseline HbA1c >/= 9%, falling by 1.22% in the high-dose group. 22 DeFronzo et al compared exenatide 10 μg or 5 μg twice daily to placebo as add on to maximal metformin therapy in 336 patients with baseline mean HbA1c of 8.2%. There were dose-dependent HbA1c reductions in the exenatide groups compared to placebo, with HbA1c change from baseline -0.78%, -0.4%, and +0.08%, respectively. 23 A similar trial compared exenatide to placebo in patients on metformin and a sulfonylurea. HbA1c reductions were similar, and again, HbA1c reductions were greater in those with higher baseline A1c values. 24 Nausea was the most common side effect reported in association with exenatide use in these trials; however, the incidence of hypoglycemia was low. In each of these trials, patients in the exenatide arms experienced mean weight reductions of 1.6 to 2.8 kg that were independent of gastrointestinal side effects.22–24
There are two reported longer-duration, open-label trial extensions with exenatide and metformin, one of two years’ and the other of three years’ duration. In the two-year extension, HbA1c reduction was maintained at 1.1% compared to 0.9% at week 30 of the initial trial. Additionally, body weight continued to steadily decline on average over the two year extension period, with a mean reduction of 4.7 kg compared to 2.1 kg after 30 weeks. 25 In the three-year extension, overall HbA1c reduction was maintained at 1.0% with a sustained weight reduction of 5.3 kg. 26
Zinman et al investigated exenatide as add on to TZD alone or with metformin in 233 patients with mean HbA1c 7.9%. Twice daily exenatide was titrated up from 5 to 10 μg twice daily after four weeks. Exenatide treatment resulted in placebo-subtracted HbA1c reduction of 0.98% and placebo-subtracted weight reduction of 1.51 kg. 27
There have been two non-inferiority studies examining exenatide versus insulin therapy as add on to sulfonylurea and metformin. In the first, exenatide was compared to glargine insulin as an adjunct to metformin and a sulfonylurea in patients with mean HbA1c 8.2%-8.3%. The dose of glargine was titrated to maintain fasting blood glucose less than 100 mg/dl. HbA1c was reduced by 1.11% in both treatment arms at 26 weeks. However, exenatide resulted in greater reduction of post-prandial glucose levels, while glargine resulted in greater reduction of fasting glucose. 28 In the second, twice daily exenatide treatment showed similar HbA1c lowering compared to biphasic aspart insulin as adjunct to sulfonylurea and metformin, with placebo-subtracted HbA1c reductions of 1.04% and 0.89%, respectively. Exenatide lead to a greater reduction in post-prandial glucose excursions. 29 In both trials the exenatide treatment arm was associated with weight reduction (-4.1 kg and -5.4 kg) compared to a weight gain associated with insulin treatment. The frequency of hypoglycemic events was similar; however, exenatide therapy was associated with fewer episodes of nocturnal hypoglycemia than was glargine.28,29 Nausea was more frequently reported in the exenatide groups. 29
A small study compared patients continued on an existing regimen of insulin plus oral medications versus those who added 10 μg twice daily exenatide to appropriately reduced insulin doses. There were no significant changes in mean HbA1c in either group. In the exenatide group, fewer patients exhibited an increase in HbA1c of greater than 0.5% over time (62% of the patients on exenatide compared to 81% of the insulin-treated patients), although patients with a longer duration of diabetes were more likely to experience glycemic deterioration when changed to exenatide. Mean weight reductions were similar to other reports. 30
A meta-analysis compared TZDs to exenatide as adjuncts to other oral agents in patients with baseline HbA1c values ranging from 7.5% to 9.9%. The weighted mean HbA1c reduction compared to baseline was greater for TZDs compared to exenatide (0.80% versus 0.60%). However, exenatide therapy was associated with mean reduced body weight of 2.74 kg versus an increase of 2.19 kg seen with TZD administration. There was no difference in rates of hypoglycemic events, but exenatide use was associated with increased gastrointestinal symptoms. 31
Exenatide LAR
Mechanism of action
Exenatide LAR, a long-acting formulation of exenatide currently in development, is composed of microspheres of exenatide and poly (lactide-coglycolide) polymeric matrix. Administration of once-weekly exenatide LAR 2.0 mg reaches a concentration shown to reduce plasma glucose after two weeks.
Efficacy in clinical studies
In a 15-week phase 2 study, exenatide LAR at doses of 0.8 mg weekly and 2.0 mg weekly were administered to 45 subjects with a mean HbA1c 8.5% on a baseline of metformin and lifestyle modification. Exenatide LAR low dose and high dose, respectively, reduced the average HbA1c by 1.4% and 1.7%, with an increase in the placebo group of 0.4% over the same time period. Only the group given higher dose exenatide LAR exhibited weight reduction (3.8 kg). Nausea and gastroenteritis were more frequent with exenatide LAR, and hypoglycemia was also more frequent in the exenatide treatment arm. Interestingly, the reduction in fasting plasma glucose with exenatide LAR was fourfold greater than had been reported in 30-week studies with 10 μg twice daily exenatide. This is thought to be due to the constant exposure to the drug conveyed by the long acting formulation. The HbA1c reduction was twice what has been associated with twice-daily exenatide therapy. 32
Safety and tolerability
The most commonly reported adverse events in patients treated with exenatide include the following: nausea (43.5%), hypoglycemia (19.6%), vomiting (12.8%), diarrhea (12.8%), feeling “jittery” (9%), dizziness (95), headache (9%), and dyspepsia (6%). 17 Nausea appears to occur in a dose-dependent fashion: this side effect may be minimized via slow dose titration of the twice daily formulation.17,18 Exenatide delays gastric emptying and is not recommended in patients with severe gastrointestinal disease or gastroparesis.17,33 Rates of hypoglycemia are higher when exenatide is administered concomitantly with sulfonylurea therapy, likely due to potentiation of the sulfonylurea effect. 17
The clearance of exenatide is predominantly through the renal system; thus, hepatic dysfunction is not expected to change its pharmacokinetic profile. 17 The use of exenatide is not recommended in patients with severe renal impairment or end-stage renal disease. 33 The pharmacokinetic profile of exenatide appears to be stable across patients of different age, race, sex, and body weight. 17
There are modest drug interactions with digoxin, lisinopril, and lovastatin. None of these medications require dosage adjustments, but close monitoring of outcome parameters associated with each drug is recommended given patient variability. 17 Also, oral agents that require rapid gastrointestinal absorption for efficacy, such as oral contraceptives and antibiotics, should be given at least an hour prior to exenatide administration. The same should be done for medications that are taken with food. 17 Finally, drugs with a narrow therapeutic range, such as warfarin or digoxin, should be given at a consistent time interval in relation to exenatide in order to maintain dose stability. 17
Exenatide is categorized as pregnancy category C. High dose exenatide in animal studies has shown teratogenic consequences affecting growth and skeletal development. There are limited data available regarding drug excretion into breast milk.
A number of case reports of acute pancreatitis in patients using exenatide have been submitted during the post-marketing period. This has prompted regulatory agencies such as the Food and Drug Administration (FDA) in the USA to endorse label warnings that recommend cessation of this agent if pancreatitis is suspected.34,35 However, a recent study investigating hospitalizations for pancreatitis in exenatide and sitagliptin cohorts, versus matched comparators, showed no increased frequency of pancreatitis with exenatide (or sitagliptin, a DPP-4 inhibitor) at 1 year of follow up. 36 The true relationship and/or frequency with which the drug contributes to pancreatitis remains unclear at present.
Liraglutide
Mechanism of action and efficacy in clinical studies
A newer GLP-1 receptor analogue, liraglutide, has been developed as a once daily medication that has a close homology to native human GLP-1. Liraglutide has a longer duration of action than exenatide, lasting 13 hours after subcutaneous administration.37,38 In a phase 2 clinical trial, liraglutide showed promising, dose-dependent HbA1c reductions of 1.27 to 1.74%. In addition, individuals in the liraglutide treatment arm had reductions in fasting plasma glucose, and the 1.9 mg dose resulted in a 1.7 kg placebo-subtracted weight reduction. Early trials of exenatide have suggested that antibodies may form in greater than 30% of patients after long-term administration;18,39 interestingly, phase III trials have suggested that liraglutide therapy results in less antibody formation than does exenatide.38,40 This is likely due to greater homology of liraglutide to the human form of GLP-1. However, antibody formation is not associated with reduced efficacy for either drug or with other adverse clinical outcome.18,37,38
The Liraglutide Effect an Action in Diabetes (LEAD) trials are a series of phase III clinical studies designed to assess the therapeutic benefits of liraglutide in the management of patients with type 2 diabetes. The LEAD-1 study was a 26-week, multi-center trial that compared liraglutide versus rosiglitazone versus placebo as add on therapy to glimepiride in patients with mean HbA1c of 8.4%-8.5%. 41 Patients treated with liraglutide had a reduction in HbA1c of 1.1% versus an increase of 0.25% in the placebo treated arm, while rosiglitazone therapy resulted in a HbA1c reduction of 0.4% from baseline. 41
LEAD -2 was a 26-week, double-blind, randomized trial that compared liraglutide 1.2 mg or 1.8 mg to placebo, as add-on to metformin alone or metformin plus glimepiride in patients with HbA1c between 7 to 10%. Both liraglutide groups had a 1.0% reduction in HbA1c as compared to placebo, and dose-dependent weight loss was noted in the liraglutide treatment arms.39,42 Gastrointestinal side effects were the most frequently reported adverse events. 42 In comparison to glimepiride, liraglutide treatment results in similar improvements in glycemic control, less hypoglycemia, and reduced body weight when administered with metformin. 42
The LEAD-3 study was a 52-week study evaluating liraglutide (1.2 mg and 1.8 mg) versus glimepiride 8 mg daily in patients with baseline HbA1c 8.3%-8.4%. After 52 weeks, the HbA1c reductions were 0.51% in the glimepiride group, 0.84% in the liraglutide 1.2 mg group, and 1.14% in the liraglutide 1.8 mg group. 43 Liraglutide monotherapy also reduced fasting and postprandial glucose levels. 39 In LEAD 4, liraglutide in combination with metformin/rosiglitazone resulted in a 1.5% HbA1c reduction compared to 0.5% lowering in the placebo treatment arm. In LEAD 5, liraglutide in combination with metformin/glimepiride yielded a 1.3% HbA1c reduction compared to 0.2% in the placebo treatment arm.39,44 Liraglutide treated patients had greater improvements in HbA1c than did those who had insulin glargine added to the oral agents. 39 Importantly, the LEAD trials found that liraglutide treatment is associated with low rates of minor hypoglycemia, and no significant increase in rates of major hypoglycemia. Rates of minor hypoglycemic events were <0.5 per patient year with liraglutide monotherapy, and 0.1-0.6 events per patient year when the drug was administered with oral agents. In LEAD 5, though, liraglutide added to metformin/sufonylurea resulted in a somewhat higher rate of minor hypoglycemic events (1.2 events per patient year). Notably, the LEAD trials found that liraglutide therapy resulted in a mean weight loss when the drug was administered either as monotherapy or in conjunction with oral agents. As seen with other GLP-1 analogues, the primary side effects of liraglutide therapy are gastrointestinal in nature. Liraglutide monotherapy has been associated with nausea in 27%-29% of subjects and diarrhea in 16%-19% of subjects. 43
Safety and tolerability
Liraglutide treatment has resulted in nausea and delayed gastric emptying in some studies. 45 Overall, use of the drug in trials thus far has not been associated with severe hypoglycemia. A stepwise dose titration has been suggested to minimize nausea and other gastrointestinal side effects. 46 Davidson et al, conducted a meta-analysis of six phase III studies and concluded that mild renal impairment (CrCl of 60-89 mL/min) had no effect on liraglutide safety or efficacy. 33
GLP-1 Agonists and non-glycemic outcomes
GLP-1 agonists may have several important non-glycemic benefits, including weight loss; small but significant decreases in systolic blood pressure; and possible preservation of pancreatic beta cell mass and/or function. Open-label extended studies of exenatide showed continued significant weight loss after 2 and 3 years of treatment. In addition, exenatide may be associated with improved lipid profiles after 3.5 years of treatment.25,26 The LEAD studies also consistently showed a reduction in body weight of around 2 kg from baseline and a mild systolic blood pressure reduction of 2 to 6 mm Hg. 39 Exenatide monotherapy in drug naïve patients with type 2 diabetes also resulted in improvements in systolic and diastolic blood pressure parameters. 21 Furthermore, both exenatide and liraglutide have been shown to increase beta cell mass in rodent models.18,37
Several studies are investigating the potential cardiovascular benefits of GLP-1 agonists. Trials designed to determine the efficacy of GLP-1 mimetics in glycemic control have noted improvements in lipid parameters such as triglycerides, total cholesterol, and HDL.26,39 Additional studies will assess the benefits of GLP-1 therapy in myocardial protection and heart failure. A study in pigs has identified exenatide as a potential agent for reducing infarction size after an acute myocardial infarction. 47 This theoretical benefit has been supported by studies suggesting that GLP-1 mediates effects on post-ischemic myocardium through a myocardial GLP-1 receptor. 48 GLP-1 infusion studies have shown improved left ventricular (LV) systolic function in dilated cardiomyopathy animal models. Phase II trials and pilot studies investigating the effects of GLP-1 infusions in humans have shown improvements in left ventricular ejection fraction (LVEF). However, at this time, future studies are needed to define the therapeutic role of GLP-1 agents in the prevention or treatment of cardiovascular disease. 48
Emerging GLP-1 analogues
Additional GLP-1 analogues in development include albiglutide, a long-acting GLP-1 mimetic engineered by genetic fusion of a DPP-4-resistant GLP-1 dimer to human albumin,49,50 and taspogluptide, a GLP-1 analogue with 93% homology to endogenous GLP and resistance to DDP-4 degradation. 51 The action profile of both agents will likely be amenable to once-weekly dosing. Another prospective GLP-1 analogue is MKC 253/GLp-1 Technosphere® proposed as an inhaled GLP-1 analogue. Results from the first human open-label, dose-escalation trial found that the administration of this analogue resulted in increases in insulin levels, as well as an increase in GLP-1 levels at some doses. 52
DPP-4 Inhibitors
Mechanisms of action
The DPP-4 enzyme circulates in soluble form in the plasma and is responsible for the inactivation of a number of hormones and peptides, including GLP-1 and GIP. Administration of agents which inhibit DPP-4 has been shown to raise levels of endogenous GLP-1 and GIP, which in turn results in a glucose-appropriate increase in insulin secretion and suppression of glucagon release. 53 Additionally, in individuals with type 2 diabetes, administration of agents which inhibit DPP-4 has been shown to increase HOMA-β and decrease the proinsulin/insulin ratio, suggestive of improvement in insulin processing. 54 Animal data suggest preservation of pancreatic beta cell mass and function mediated by DPP-4 inhibition; however, no comparable data in humans exists. 55 Unlike the GLP-1 analogues, DPP-4 inhibitors have not been shown to increase satiety, slow gastric emptying, or reduce food intake. 56 The DPP-4 inhibitors sitagliptin and vildagliptin are currently available for the management of type 2 diabetes; however, vildagliptin is not presently available in the USA.
Sitagliptin
Sitagliptin is a DPP-4 inhibitor currently approved for use in Europe, USA, and many other countries. Sitagliptin doses of 50 mg and 100 mg inhibit DPP-4 activity by 80% over 12 and 24 hours, respectively. This is the level of inhibition at which near maximal glucose lowering is seen. 57
Efficacy in clinical studies
A total of 11 large trials of sitagliptin as monotherapy or as add-on therapy have been published to date. In 2006, there were two similar studies of sitagliptin monotherapy. The two studies enrolled 741 and 521 patients for 24 and 18 weeks, each randomizing patients to sitagliptin 100 mg, sitagliptin 200 mg, or placebo. Placebo subtracted HbA1c reductions ranged from 0.48% to 0.94%, with no clear dose response pattern. Patients with higher baseline HbA1c (>9%) had modestly better reduction in HbA1c, at just over 1%. Fasting glucose, postprandial glucose, HOMA-β, and insulin/proinsulin ratios were also improved in the sitagliptin groups. These trials did not show an increased incidence of hypoglycemia in the sitagliptin groups, nor was there a significant change in weight. As there was no demonstrable additional glucose lowering benefit seen with the 200 mg dose, subsequent trials utilized 100 mg daily as the maximum daily dose.58,59
Goldstein et al conducted a randomized controlled trial of sitagliptin versus metformin versus combination therapy with the two drugs. A total of 1091 patients uncontrolled on diet and exercise, with HbA1c 7.5%-11%, were randomized to one of six groups: placebo, sitagliptin 100 mg daily with metformin 2000 mg daily (S100/M2000), sitagliptin 100 mg daily with metformin 1000 mg daily (S100/M1000), metformin 2000 mg daily (M2000), metformin 1000 mg daily (M1000), and sitagliptin 100 mg daily (S100). (All metformin groups received twice daily divided doses). Placebo-subtracted HbA1c reductions were as follows: S100/M2000 -2.07%, S100/M1000 -1.5%, M2000 -1.3%, M1000 -1.0%, S100 -0.8%. Individuals randomized to a combination regimen had significantly greater HbA1c reduction than did monotherapy groups. The incidence of gastrointestinal adverse events was similar across groups, and rates of hypoglycemia were low across treatment groups and similar to placebo. 60 In addition, there have been three large trials of sitagliptin as adjunctive therapy to metformin in patients with inadequate glucose control on metformin alone: two placebo-controlled studies and one with an active control. The placebo-controlled trials enrolled 701 and 190 patients and lasted 24 and 30 weeks, respectively. They differed slightly in baseline HbA1c's, with the first enrolling patients on metformin with HbA1c between 7% and 10% and the second with HbA1c between 8% and 11%. Placebo subtracted HbA1c reductions seen in the sitagliptin groups were 0.65% and 1.0%. No increased hypoglycemia or weight gain was seen in either of the sitagliptin groups, and markers of beta cell function, when measured, were significantly improved as well.59,61 In the active control trial, sitagliptin 100 mg was compared to glipizide 5 to 20 mg for 52 weeks in 1172 patients with HbA1c between 6.5 and 10% on metformin monotherapy. Sitagliptin was found to be non-inferior to glipizide, with a mean HbA1c reduction of 0.67% in both groups. In addition, hypoglycemia was less common in the sitagliptin group than the glipizide group, and the sitagliptin group lost a mean of 1.5 kg body weight, as compared with a 1.1 kg gain in the glipizide group. 62
Two other adjunctive therapy trials combined sitagliptin with glimepiride or pioglitazone. The first was a trial of 441 patients on glimepiride alone or glimepiride plus metformin, with baseline HbA1c of 7.5 to 10.5%, randomized to receive additional therapy with either sitagliptin 100 mg daily or placebo for 24 weeks. In the sitagliptin group, the HbA1c was reduced by 0.74% relative to placebo. Significant improvements in fasting plasma glucose, post-prandial glucose, and HOMA-β were seen as well. In this study, unlike the studies described above, there was an increased incidence of hypoglycemia and a modest weight gain (0.8 kg versus -0.4 kg) with sitagliptin relative to placebo. The authors speculate that this is related to potentiation of the sulfonylurea effect. 63 The second study added sitagliptin or placebo to pioglitazone as adjunctive therapy in 353 patients with baseline HbA1c 7% to 10%. After 24 weeks, a placebo-subtracted HbA1c reduction of 0.7% was seen in the active therapy group, without an increase in adverse events. 64
A small, short duration, head-to-head study comparing sitagliptin to exenatide showed greater glucose-lowering with exenatide. The study was conducted in metformin-treated patients with type 2 diabetes and mean baseline HbA1c 8.5%. Patients were randomized to exenatide (5 μg BID for 1 week, then 10 μg BID for 1 week) or sitagliptin (100 mg daily) for 2 weeks. After 2 weeks, patients crossed-over to the alternate therapy. After the first treatment period, 2-h PPG was significantly lower with exenatide than with sitagliptin: 133 mg/dl versus 208 mg/dl. After cross-over, the patients switched from sitagliptin to exenatide showed improvement in 2-hr PPG, while those switched from exenatide to sitagliptin had worsening. Investigators also found reduced total caloric intake in the exenatide group compared to the sitagliptin group (-134 kcal/day vs. +130 kcal/day) as well as slowed gastric emptying as measured by labeled acetaminophen study. 56
Safety and tolerability
Seventy nine percent of the administered sitagliptin dose is excreted unchanged in the urine via active tubular secretion.65,66 The drug does not induce the cytochrome P450 system and is not expected to interact with drugs metabolized through this pathway. 65 Drug interactions have not been seen in studies evaluating combinations with glyburide, metformin, rosiglitazone, and pioglitazone.64,67–69 Sitagliptin has not been studied in combination with insulin.
Sitagliptin is safe for use in patients with renal insufficiency, although the dose should be reduced to 50 mg daily for creatinine clearance 30 to <50 ml/min and to 25 mg daily for creatinine clearance <30 ml/min.70,71 Drug metabolism does not differ in obese as compared to lean subjects. 66 Sitagliptin has been studied in patients with diverse ethnic backgrounds, including Japanese, Korean, Chinese, and Indian subjects, with apparent similar activity in all of these groups.72,73
There have been postmarketing reports of a few serious hypersensitivity reactions, including angioedema, anaphylaxis, and exfoliative skin conditions, in patients treated with sitagliptin. Some of these events occurred shortly after initial drug administration. Previous serious hypersensitivity reaction is currently the only contraindication to the use of sitagliptin. 74
Vildagliptin
Vildagliptin is an inhibitor of DPP-4 currently available in Europe and many other countries, although approval in the US is still pending. Vildagliptin has been shown to suppress endogenous glucose production by increasing circulating incretin levels; it also appears to enhance measures of islet cell function in patients with both type 2 diabetes and impaired glucose tolerance.75–77 Interestingly, Azuma et al showed that vildagliptin improves glucose metabolism in peripheral tissues, as measured by an insulin infusion study. Improvement in peripheral glucose utilization is a novel finding for drugs targeting the incretin system–-the authors speculate that it may be a direct effect of GLP-1 or GIP on glucose uptake. 78
Efficacy in clinical studies
There have been 14 large trials examining vildagliptin in patients with type 2 diabetes. Several studies have evaluated its role as monotherapy in drug-naïve patients and to determine the appropriate therapeutic dosing strategy. In the first study, 98 drug-naïve patients were randomized to vildagliptin 25 mg bid versus placebo. Mean placebo-subtracted changes in HbA1c were 0.6% and 1.2%, in patients with baseline HbA1c levels of 8 or 9.5%, respectively. Improvement in beta cell function in the vildagliptin group was suggested by improvements in fasting glucose, corrected insulin response at peak glucose, and mean prandial c-peptide. 79 In the second trial, 354 drug-naïve patients were randomized to placebo versus vildagliptin 50 mg daily versus 50 mg twice daily versus 100 mg daily. Improvement in HbA1c was seen in all dosage groups, with placebo-subtracted reductions as follows: 50 mg daily 0.5%, 50 mg twice daily 0.7%, and 100 mg daily 0.9%. No increase in adverse events, hypoglycemia, or weight gain was seen. 80 Similar results were seen in a 24-week trial of 632 drug-naïve patients with average baseline HbA1c of 8.4%. A more modest reduction in HbA1c (0.3%) was noted in a 52-week trial of patients with a lower baseline HbA1c of 6.2 to 7.5%.81,82 Vildagliptin has undergone noninferiority comparisons with metformin, pioglitazone, acarbose, and rosiglitazone. In the two trials comparing vildagliptin with metformin, investigators reported somewhat different outcomes. In the first, vildagliptin 100 mg daily was found to be non-inferior to metformin 2000 mg daily, with both groups demonstrating HbA1c reductions of 1.0%. 83 However, in a second trial, metformin 2000 mg daily showed statistically significantly better reduction in HbA1c than vildagliptin 100 mg daily (1.5% versus 1.0%). 84 In another trial, Rosenstock et al compared vildagliptin 100 mg daily vs. pioglitazone 30 mg daily vs. combination therapy with vildagliptin/pioglitazone 100/30 mg or 50/15 mg in drug-naïve patients in a 24-week trial. HbA1c reductions were 1.1%, 1.4%, 1.9%, and 1.7%, respectively. Both combination therapies were more effective in improving glycemic control than was therapy with either single agent. Peripheral edema was most frequent in patients receiving pioglitazone monotherapy (9.3%) and least frequent in the low-dose combination group (3.5%). 85
In a pioglitazone non-inferiority trial, vildagliptin 100 mg daily showed similar reduction in HbA1c at 24 weeks when compared with pioglitazone 30 mg daily and was non-inferior by statistical comparison. There was significantly more weight gain in the pioglitazone group (+1.9 kg versus -1.6 kg with vildagliptin). 86 When compared with acarbose, vildagliptin had similar efficacy but was better tolerated. 87 Finally, vildagliptin 100 mg daily was compared with rosiglitazone 8 mg daily in drug-naïve patients and was shown to be non-inferior with similar HbA1c reduction. In this trial, patients treated with vildagliptin experienced reductions in total and LDL cholesterol levels, while those treated with pioglitazone had increases in HDL cholesterol. 85
Adjunctive therapy trials with vildagliptin have included combinations with insulin, pioglitazone, and metformin. HbA1c reductions were similar overall to those seen in the trials described above, and no increase in hypoglycemia or weight gain was seen in the vildagliptin groups.88–90 Available data does not appear to show that vildagliptin alters gastric emptying or the rate of entry of ingested glucose into the systemic circulation in humans. 91
Safety and tolerability
Vildagliptin is similar to sitagliptin in that it is generally well-tolerated and does not appear to cause significant hypoglycemia or weight gain. 92 Rare cases of hepatic dysfunction have been reported, and vildagliptin is not recommended for use in those with moderate to severe hepatic dysfunction. 93 Skin blistering was noted in non-clinical toxicology studies with primates, although this has not been reported in human studies at recommended therapeutic dosages. 94 More studies are needed to examine its potential immunomodulatory effects as well as its use in patients with renal insufficiency.
Emerging DPP-4 inhibitors
A variety of other DPP-4 inhibitors are in either early or late stages of drug development. Those most likely to become available in the near future include saxagliptin and alogliptin. A monotherapy trial with saxagliptin at various doses evaluated 338 drug-naïve patients with type 2 diabetes and found placebo subtracted HbA1c reductions of 0.45%-0.63% across all arms. Similar to other drugs in this class, there was no effect on weight or additional noted adverse events. 95 Another trial evaluated saxagliptin 2.5 mg or 5 mg daily versus placebo as adjunctive therapy to a thiazolidinedione in patients with HbA1c 7 to 10.5%. The patients treated with saxagliptin had a placebo-subtracted HbA1c reduction of 0.36% in the 2.5 mg group and 0.64% in the 5 mg group. Improvements were also seen in fasting and postprandial glucose. Hypoglycemia and adverse event rates were similar in frequency to placebo. 96
A 26-week, alogliptin monotherapy trial was conducted in 329 diabetic patients with mean HbA1c 7.9% on diet and exercise. Participants were randomized to alogliptin 12.5 mg/day, alogliptin 25 mg/day, or placebo. Both alogliptin doses produced significant reductions in HbA1c as compared to placebo (0.56% and 0.59% versus 0.02%). Hypoglycemia and weight gain were not seen. 97 A trial of alogliptin 12.5 mg or 25 mg daily versus placebo as adjunctive therapy with metformin was conducted in 527 patients with mean HbA1c 7.9%. The alogliptin groups showed significantly greater reductions in HbA1c than placebo (0.6% for both doses versus 0.1% for placebo). No significant increase in weight, hypoglycemic events, or gastrointestinal side effects was seen. 98 Finally, a randomized trial added alogliptin 12.5 mg or 25 mg versus placebo to established insulin therapy in patients with inadequate glucose control (mean baseline HbA1c 9.3%). The alogliptin groups had greater efficacy than placebo, with a placebo subtracted reduction in HbA1c of 0.5 and 0.58% for the respective doses. No between-group differences in weight were seen, and there was a similar overall incidence of hypoglycemia. 99
One of the major benefits of the DPP-4 inhibitor class is that the medications are generally well-tolerated. Reported side effects are similar for sitagliptin and vildagliptin and include headache, which is more frequent with vildagliptin. 100 Dose reduction is recommended in patients with moderate to severe renal failure.70,101 A 2009 Cochrane review compiled safety data from 25 trials of sitagliptin and vildagliptin. They reported a statistically significant increase in all-cause infections (including nasopharyngitis, upper respiratory tract infection, urinary tract infection, and other) in the sitagliptin group, with a relative risk of 1.15. The trend did not reach statistical significance for vildagliptin. 100 In general, trials have not shown increased rates of hypoglycemia attributable to DPP-4 inhibitors, and weight trends have been neutral. DPP-4 inhibitors have not been studied in pregnant or lactating women.
Amylin Analogue
Amylin hormone
In 1987, a 37-amino acid pancreatic neurohormone called amylin was discovered. It is secreted post-prandially by the beta cell, along with insulin. Amylin complements insulin action in mealtime glucose control by decreasing glucagon secretion, slowing gastric emptying, and enhancing satiety.102,103 Amylin receptors are located in distinct areas of the brain; the hormone's effects in the postrema and dorsal motor nucleus of the vagus are likely involved in satiety and food intake. Under normal conditions, amylin is secreted in high frequency pulses every 4-6 minutes. 102 Individuals with type 1 diabetes have deficiency of amylin secretion, thought to be related to beta cell destruction. However, individuals with type 2 diabetes have initially elevated amylin levels that decline as the disease progresses, mirroring the pattern of insulin secretion in the disease.103,104
Pramlintide
Mechanism of action
Pramlintide acetate i s a commercially available synthetic analog of amylin that has physiologic effects similar to those of the endogenous hormone. Administered as a pre-meal subcutaneous injection, it has been shown to have a bioavailability of approximately 38 to 40%. It achieves a maximum level at 20 minutes and lasts 3 hours after administration. The elimination halflife is approximately 20-45 minutes. Pramlintide is currently approved as an adjunct to mealtime insulin in patients with uncontrolled type 1 or type 2 diabetes. Mealtime dosing starts at 60 μg in patients with type 2 diabetes with titration up to a maximal maintenance dose of 120 μg, while a starting mealtime dose of 15 μg in patients with type 1 diabetes is titrated up to a maximal maintenance dose of 60 μg. 105
Efficacy in clinical studies
In a randomized, multicenter study, 538 insulin-treated subjects with type 2 diabetes were given pramlintide 30 μg, 75 μg, 150 μg, or placebo with meals. At 52 weeks, mean HbA1c reduction was 0.6% in those treated with pramlintide 150 μg as compared to 0.1% in the placebo group. 106 A second large multicenter study randomized 656 patients with type 2 diabetes to receive pramlintide 90 μg BID, 120 μg BID, 60 μg TID, or placebo, along with existing doses of insulin and oral medications. Participants in the BID arms received an additional placebo injection. At 52 weeks, there was significant improvement in HbA1c in all pramlintide arms. The pramlintide groups achieved up to a threefold greater proportion of patients with HbA1c <7% and an almost twofold greater proportion of patients with HbA1c <8%. In addition, pramlintide 120 μg BID treated group achieved a -1.4 kg vs +0.7 kg weight change compared with placebo at week 52. P < 0.05). 107
Two placebo-controlled studies have specifically looked at the role of pramlintide as an adjunct to insulin for treatment of overweight and obese patients with type 2 diabetes. In the first, those randomized to pramlintide 120 μg BID achieved a placebo corrected HbA1c reduction of 0.41% at 26 weeks of therapy. 108 Similar HbA1c reductions were seen in the second trial, which also revealed a pramlintide-related weight reduction of 2.0 kg compared to placebo. 102 Lastly, pramlintide was examined in a multiethnic trial, which enrolled Whites, Blacks, and Hispanics. In this study, similar HbA1c reductions were shown across ethnic groups, suggesting that pramlintide's effects appear to be generalizable. 109
Safety and tolerability
Available safety data for pramlintide indicate that the most common side effects are nausea, anorexia, and headaches, with incidences of ≥10%.107,110 These effects appeared to be dose-related and were of mild-to-moderate intensity. 102 Pramlintide seems to be generally well tolerated, and, to date, there is no evidence of increased cardiovascular, pulmonary, hepatic, renal, or idiosyncratic drug-related adverse events.104,107,110 Pramlintide is contraindicated in patients with hypersensitivity to pramlintide or metacresol, gastroparesis, or hypoglycemia unawareness. It is recommended that prandial insulin doses be decreased in patients starting pramlintide in order to reduce the likelihood of subsequent hypoglycemia, particularly in patients with type 1 diabetes.
Dopamine Agonist
Bromocriptine mesylate
Mechanism of Action
Bromocriptine mesylate is a medication recently approved by the United States FDA for the management of type 2 diabetes mellitus as an adjunct to diet and exercise. Bromocriptine mesylate, an ergot derivative, is a sympatholytic dopamine D2 receptor agonist that can exert inhibitory effects on serotonin turnover in the central nervous system. 111 This medication decreases blood glucose levels via central signaling. Current evidence suggests that this medication reverses metabolic abnormalities associated with insulin resistance by resetting hypothalamic circadian organization of monoamine neuronal activities. However, the specific mechanism by which bromocriptine mesylate improves glycemic control is not clearly elucidated.33,104 Bromocriptine mesylate is a new quick-release oral formulation of bromocriptine. When administered orally, approximately 65%-95% of the dose is absorbed. The medication is metabolized in the liver by CYP3A4, and in the fasting state, the time to maximum plasma concentration is 53 minutes. It is excreted in the bile. There are no data available regarding the pharmacokinetics of this medication in renal impairment, hepatic impairment, or the pediatric population. Bromocriptine mesylate is considered pregnancy category B and is contraindicated in mothers who are nursing. It is recommended that patients take this medication within two hours after waking. Recommended doses start at 0.8 mg daily, increased weekly by one tablet, until a maximal tolerated daily dose of 1.6 to 4.8 mg is achieved. 35
Efficacy in clinical studies
The efficacy of bromocriptine mesylate has been documented in several clinical trials, including a randomized, controlled trial evaluating its use as monotherapy in patients with type 2 diabetes. In this study, a total of 159 overweight patients with type 2 diabetes and HbA1c levels between 7.5%-11% were randomized to the active drug vs placebo for a total of 24 weeks. At completion of the study, patients randomized to active therapy achieved a placebo-subtracted HbA1c reduction of 0.4%. Further, two 24-week clinical trials enrolled patients with inadequately controlled diabetes on sulfonylurea to the addition of bromocriptine mesylate versus placebo. In both studies, patients randomized to the active drug plus sulfonylurea achieved reductions in placebo-subtracted HbA1c of 0.5% and 0.6%. Similar efficacy has been documented in other trials of bromocriptine mesylate as add-on therapy in patients with uncontrolled type 2 diabetes on a baseline of 1-2 oral medications. 35
Safety and tolerability
The most common adverse reactions experienced by patients treated with bromocriptine mesylate in clinical trials were nausea, fatigue, dizziness, vomiting, and headache, reported in ≥5% of participants. The medication is contraindicated in patients with hypersensitivity reactions to ergot derivatives, in nursing mothers, and in patients with syncopal migraines. Further, this class of medication might cause orthostatic hypotension or somnolence, may precipitate psychosis, and can cause interactions with dopamine antagonists. 35
Emerging Drug classes for Glucose Lowering
Sodium glucose transporter-2 (SGLT2) inhibitors
The sodium glucose transporters are a family of membrane proteins found in the intestinal epithelium and the proximal renal tubules that actively transport various molecules, including glucose, amino acids, vitamins, osmolytes, and ions, across the cell membranes.112–114 SGLT2 is a specific SGLT protein that is expressed in the renal cortex. Its activity accounts for 90% of glucose reabsorption in the kidney.112,114 SGLT2 has significant structural affinity with glucose transporter 2(GLUT)-2, a well-known glucose transport protein. Natural mutations in SGLT2 have been reported and are noted to cause increased glucose excretion. This observation served as the foundation for the development of selective inhibitors of SGLT2, which, in theory, would lower blood glucose by preventing renal glucose reabsorption. 115
Two SGLT2 inhibitors are currently under investigation: dapagliflozin and sergliflozin. Dapagliflozin has 1200-fold selectivity for SGLT2, with similar inhibitory potencies in rat and human SGLT2 studies. When administered to diabetic rats, this medication produced dose-dependent glucosuria, enhanced glucose tolerance, and decreased hyperglycemia.114–116 Sergliflozin is a highly selective inhibitor of SGLT2. In animal models, oral administration of sergliflozin decreased plasma glucose by increasing urinary glucose excretion in a dose-dependent manner. In glucose tolerance tests, sergliflozin exhibited glucose-lowering effects independent of insulin levels. In addition, in animal models, sergliflozin improved postprandial hyperglycemia and reduced levels of glycated hemoglobin and plasma glucose. Sergliflozin did not affect body weight, food intake, or electrolyte balance.114,117,118 An additional agent, remogliflozin etabonate, has also shown promise in animal studies. 119
Interleukin-1 receptor antagonist
The interleukin-1 receptor antagonist, a competitive inhibitor of interleukin-1 at the type I receptor, protects humans beta cells from glucose-induced apoptosis. As patients with diabetes mellitus type 2 have decreased pancreatic islet cell expression of the interleukin-1-receptor antagonist, studies have been conducted to assess the potential role of interleukin-1 receptor antagonist therapy in diabetes management. In 2007, a randomized, placebo controlled, double-blind, parallel-group trial involving 70 patients was conducted using the recombinant human interleukin-1 receptor antagonist anakinra in patients with type 2 diabetes. At the end of the trial (13 weeks post randomization), the group randomized to anakinra had a 0.46% lower glycated hemoglobin level than did the group receiving placebo (P = 0.03). In addition, the medication was well tolerated without apparent serious adverse events. 120
Conclusion
The number of individuals affected by type 2 diabetes continues to increase worldwide. Fortunately, our evolving understanding of type 2 diabetes pathophysiology serves as the foundation for the development of agents which will utilize novel mechanisms in the management of hyperglycemia. As discussed in this article, the availability of newer agents such as the GLP-1 analogues, DPP-4 inhibitors, and pramlintide have already provided additional options for improving glycemic control. These newer classes may provide benefits not seen with many traditionally used antihyperglycemic agents, such as weight reduction or weight neutrality and a minimal risk for hypoglycemia. However, the well-described gastrointestinal side effects and the need for injections of GLP-1 analogues and pramlintide may limit widespread adoption of these classes. It is likely, though, that long acting formulations of GLP-1 analogues requiring less frequent dosing will be appealing to patients and prescribers alike. Studies of the newer classes of agents, particularly the incretin-based therapies, do suggest that the drugs’ mechanisms of action complement those of traditionally used diabetes medications–-this is of great importance, as it is likely that individuals with type 2 diabetes will require several types of glucose lowering medications to appropriately gain and maintain adequate glycemic control. More information regarding safe and effective multidrug combinations will be highly anticipated, particularly with respect to combinations of these newer medications with insulin as well as the use of combined incretin-based therapies. It may also be important to evaluate the potential glucose-lowering effects of drugs designed for other purposes, as has been demonstrated in studies of the bile acid sequestrant colesevelam hydrochloride. 121
Additional, likely long-term trials will be needed to determine if preliminary data suggesting beta cell preservation by some of these agents will be borne out in clinical practice. Furthermore, the need to adequately assess the cardiovascular safety of all diabetes medications has been an area of recent major emphasis. Suggestions of potential cardiovascular benefits conveyed by incretin-based and other new therapies due to effects such as weight loss/stability or improvements in blood pressure and lipids will need to be evaluated via appropriately designed clinical trials. If these benefits are substantiated, the expense of preferential use of these newer medications may be justified. It is also reasonable to expect that combinations of several drugs not likely to cause hypoglycemia (as is the case with many of these newer agents) may in fact facilitate the achievement of HbA1c goals. In any event, the ever expanding armamentarium of agents available for the management of type 2 diabetes will permit increased individualization of (and hopefully adherence to) glucose-lowering therapies.
Disclosures
The authors report no conflicts of interest.
Footnotes
Acknowledgements
Dr. Corsino is supported by NIH Training Grant T32 DK007012-30S1 at Duke University Medical Center. Drs. Cox and Rowell are supported by NIH Training Grant T32-DK007012-31 at Duke University Medical Center.