
Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is a potentially life-threatening acquired clonal stem cell disorder resulting in hemolysis and thrombosis. A somatic mutation in the phosphatidylinositol glycan A gene in hematopoietic stem cells results in a deficiency of the complement regulatory proteins, CD55 and CD59. There is a consequent increased sensitivity to complement mediated lysis. Eculizumab is a recombinant, humanized monoclonal antibody directed against C5 of the complement system. It blocks terminal complement activation and the formation of the membrane attack complex. It has been demonstrated in 3 clinical studies that this blockade of the terminal complement system effectively and significantly prevents intravascular hemolysis thereby abolishing or reducing the need for transfusions, reduces thromboses, improves quality of life and appears to prevent renal damage and improve pulmonary pressures. The drug is very well tolerated with few safety concerns. Case reports of the use of eculizumab in pregnancy, cold agglutinin disease and atypical hemolytic syndrome are also described.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is characterised by intravascular hemolysis, venous thrombosis and is associated with aplastic anemia and other bone marrow failure syndromes. 1 PNH arises through a somatic mutation of the phosphatidylinositol glycan A (PIG-A) gene in bone marrow stem cells. The PIG-A gene codes for a protein, which is critical in the catalysis of the first step of glycosylphosphati-dylinositol (GPI) biosynthesis. 2 A single mutation disrupting GPI biosynthesis results in a deficiency of all GPI-anchored proteins on the cell membrane and in the PNH phenotype.3–5
Under normal circumstances, PIG-A deficient cells are rapidly eliminated by autologous complement activation. 6 There is conclusive evidence that PNH only develops in individuals who have a predisposition to the development or, more likely, the expansion of the GPI-deficient hematopoietic clones. 7 This predisposition is probably the presence of an underlying bone marrow failure, usually aplastic anemia, either preceding or co-existent with the diagnosis of PNH.8–10 In PNH, it appears that normal hematopoiesis is suppressed by the immune system, presumably either directly or indirectly through one or more GPI-linked proteins, and that this attack spares the GPI-deficient PNH clone. Thus, in an environment where there is intense pressure for hematopoiesis the PNH clone is driven to produce mature hematopoietic cells and expands to fill the void left by the aplastic process.
Among the deficient proteins are the complement regulatory proteins CD55 (previously known as Decay Accelerating Factor) and CD59 (previously known as Membrane Inhibitor of Reactive Lysis). The resulting increased complement sensitivity of PNH cells leads to intravascular hemolysis and thrombosis. 11 Ongoing hemolysis and/or insufficient hematopoiesis often result in transfusion dependence. Hemolysis in patients with PNH can be monitored by levels of the enzyme lactate dehydrogenase (LDH). LDH is a standard biochemical measure of intravascular hemolysis and probably the most sensitive. Levels are frequently elevated in patients with PNH, exceeding 20 times the upper limit of normal during severe paroxysms.12–15
CD59, a GPI-linked protein, prevents terminal complement components from forming the hemolytic membrane pore, C5b-9. It reacts with C5-8 and with C5-9 and inhibits the insertion of C9 into cell membranes.16–18 It is widely accepted that the absence of CD59 from PNH red cells is the cause of their increased sensitivity to complement mediated lysis.6,19–24
The platelets in PNH patients also lack GPI-anchored proteins. The absence of CD59 renders platelets susceptible to attack by complement. When this occurs, the platelets undergo morphological changes which result in the release of vesiculated membrane attack complex. These vesicles or micro-particles are very procoagulant in vitro and are present at significantly elevated levels in the blood of patients with PNH. It is therefore possible that the absence of CD59 from platelets leads to thrombin generation and increased thrombotic risk.22,25,26
Clinical Consequences
The consequence of deficiency of GPI-linked proteins in PNH is brisk intravascular hemolysis leading to hemoglobinuria, dysphagia, recurrent abdominal pain, severe lethargy and erectile failure. PNH is a chronic condition, frequently affecting young individuals, that may persist for many years and which often presents clinicians with difficult management problems. The symptoms associated with ongoing hemolysis and/or insufficient hematopoiesis have a major impact on the patient's well-being. The acute exacerbations can occur either regularly or unpredictably, and have a further adverse impact on quality of life. Anemia and the need for transfusions to sustain hemoglobin levels occur frequently.
The most feared complication of PNH is venous thrombosis which occurs in approximately 50% of patients with hemolytic disease and is the cause of death in at least one-third.1,27,28 There is a predilection for the intra-abdominal and cerebral veins although deep vein thrombosis is a common presentation.
During severe bouts of hemolysis (paroxysms), hemoglobin escapes into the plasma, dimerizes and can saturate biochemical systems in place to remove it, resulting in hemoglobinuria and free plasma hemoglobin. 15 Cell-free plasma hemoglobin consumes nitric oxide (NO) rapidly. 29 In addition to hemoglobin decompartmentalization and NO scavenging, 30 hemolysis also releases erythrocyte arginase, an enzyme that converts L-arginine, the substrate for NO synthesis, to ornithine, thereby further reducing the systemic availability of NO.31–34 NO is a regulator of smooth muscle tone and platelet activation, and reductions in nitric oxide plasma levels lead to smooth muscle dystonias, including hypertension, gastrointestinal contractions, and erectile dysfunction, as well as clot formation.35–38 The best protection against NO depletion and thereby these consequences would be to stop hemolysis.
Targeting Complement
Complement is an important humoral innate immune defence system against invading microorganisms. Complement activation is a complex cascade leading to the production of bioactive anaphylatoxins, chemotaxins, opsonic fragments and the potentially lytic membrane attack complex (MAC) on the surface of target membranes.39–42 There are three pathways which activate the cascade (Fig. 1)–-the classical (initiated by antigen/antibody complex), alternative (initiated by microbial membranes or immune complexes) and lectin pathways.
These three pathways all converge to cleave C5, by either the classical or the alternative C5 convertase, into C5a, a potent anaphylatoxin, and C5b. C5a possesses various proinflammatory properties and enhances the procoagulant activity of neutrophils. 43 C5b is the initial molecule of terminal complement and binds C6 then C7 and then C8. The terminal complement components C6, C7, C8 and C9 are sequentially activated.39,40 C5b-8 forms the scaffold for C9 molecules which bind to each other to form the MAC (membrane attack complex, also called the terminal complement complex). The MAC forms pores in the cell membrane and is responsible for the characteristic intravascular hemolysis in PNH.
Eculizumab
To prevent complement-mediated diseases or adverse effects one strategy is to block the complement cascade. Individuals with inherited deficiency of any of the complement molecules prior to C5 are vulnerable to both pyogenic organisms and to autoimmune disorders. In contrast, deficiency of any of the molecules after C5, remarkably, has little abnormal phenotype. The only apparent adverse effect is an increased risk of infection by encapsulated organisms, particularly Neisseria meningitidis, although terminal complement deficient individuals appear to have a lower mortality from such infection when compared to complement-replete individuals.44–46
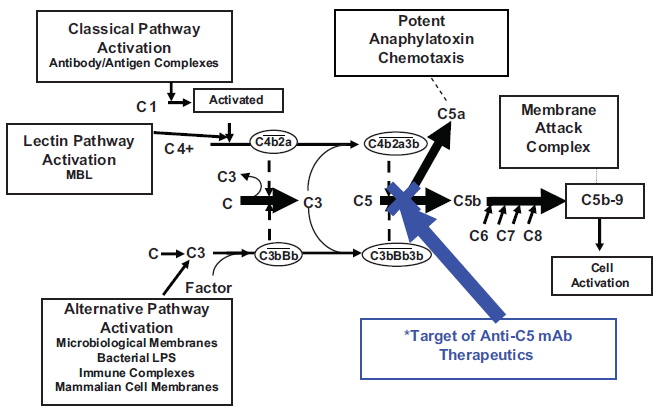
The complement cascade. The 3 pathways activating this cascade are demonstrated (Classical, Alternative and Lectin). All 3 pathways converge at C5, following which i) the membrane attack complex (MAC) is formed that results in lysis of the cell through release of C5b which subsequently binds C7 and C8; C9 molecules then bind together to form the MAC or ii) C5a, a potent anaphylatoxin, is released. in PNH, the MAC is formed readily due to loss or reduction of the terminal complement inhibitor, CD59. Prevention of formation of the MAC may be achieved by blocking terminal complement. The site of blockage of eculizumab (see later) is demonstrated.
The demonstration that a monoclonal antibody, raised against human C5, blocked C5 cleavage in vitro raised the prospect of inhibiting complement in vivo using such reagents.47,48 Eculizumab (h5G1.1-mAb, Soliris, Alexion Pharmaceuticals) is a recombinant humanized chimeric monoclonal antibody that specifically targets the complement protein C5 and prevents its cleavage. 49 All patients who receive this drug should be vaccinated against Neisseria meningitidis.
Results from a 12-week open-label study of eculizumab in patients with hemolytic PNH demonstrated a dramatic reduction in hemolysis and a concomitant increase in the proportion of PNH type III red blood cells. 12 The drug was given by intravenous infusion over 30 minutes and was well tolerated. Eculizumab was administered as 4 weekly 600 mg doses (induction regimen), followed by 900 mg doses given every other week (maintenance regimen), starting 1 week after the induction (week 5), and is the dosing schedule which has been used for all subsequent studies. In addition, this initial study showed a marked decrease in the rates of paroxysms and blood transfusions and an improvement in quality of life. An evaluation of the results of a 1-year follow-up study designed to assess the long-term efficacy and safety of eculizumab in patients with PNH was performed. 50 The immediate reduction in LDH levels observed in all patients during the acute-phase study was maintained during the extension study. The dramatic reduction in hemolysis during eculizumab treatment was demonstrated in both non-cytopenic (platelet count ≥ 150 × 109/L) and cytopenic (platelet count < 150 × 109/L) patient populations.
A statistically significant reduction in the rate of packed red blood cell transfusion was maintained during the extension study compared with the rate of transfusion before eculizumab therapy. Mean transfusion rates decreased from 2.1 units per patient each month during the 1-year period before treatment to 0.5 units per patient each month during the total 64-week treatment period. Similarly, median transfusion rates decreased from 1.8 units per patient each month before treatment to 0.3 units per patient each month during the total 64 weeks of therapy (P = 0.001). Importantly, 3 patients with hypoplasia who received transfusions during the acute-phase study were subsequently treated with erythropoietin during the extension study. Two of the 3 patients responded with an increase in reticulocyte counts and a further reduction in transfusion requirements. These data suggest that erythropoietin therapy in PNH patients with hypoplasia increases erythropoiesis and that concomitant eculizumab therapy protects against increased hemolysis, with a resultant improvement in response. 51
One patient had a breakthrough of serum hemolytic activity during the acute-phase study, and another patient broke through early in the extension study. Pharmacokinetic analysis of both breakthrough patients demonstrated a simultaneous decrease in eculizumab level below that required to completely block complement activity (≥35 μg/mL). 49 Adjustment of the eculizumab dosing interval in these 2 patients from every 14 days to every 12 days (as allowed within the trial protocol) successfully sustained trough levels of eculizumab higher than 35 μg/mL and consistently blocked serum hemolytic activity for the remainder of the extension study. Since the end of the trial, these patients have changed from 900 mg dosing every 12 days to 1200 mg every 14 days for convenient scheduling. Blockade appears to be maintained. 52 LDH levels can be used as an accurate measure of intravascular hemolysis in PNH, and they provide evidence that effective complement blockade during eculizumab therapy can be determined by monitoring levels of this enzyme.
These observations were extended to include clinical improvements in symptoms not captured by the QLQ-C30 (Quality of Life Questionnaire-Core 30) instrument attributed specifically to hemolysis including hemoglobinuria, abdominal pain, dysphagia and erectile dysfunction. 53 Clinical assessment of these PNH symptoms showed complete resolution, or at least dramatic improvement, during eculizumab treatment. Patients in the original 11 patient eculizumab trial that reported one or more of these symptoms prior to treatment all responded with dramatic improvements in clinical signs and symptoms. 50
Following the pilot study, a Phase III randomized, placebo-controlled trial (Transfusion Reduction Efficacy and Safety Clinical Investigation, a Randomized, Multicenter, Double-Blind, Placebo-Controlled, Using Eculizumab in Paroxysmal Nocturnal Hemoglobinuria–-TRIUMPH) was conducted and results from this study were reported in 2006. 54 Entry criteria included requiring 4 or more transfusions in the preceding 12 months and a platelet count greater than 100 × 109/L. The study investigated whether eculizumab improved anemia, as measured by stabilized hemoglobin levels and reduced transfusion requirements in transfusion-dependent patients with PNH during 6 months of treatment. The effect of eculizumab on chronic intravascular hemolysis was demonstrated by an immediate and sustained decrease in lactate dehydrogenase levels. At the end of the treatment period, 49% of patients in the eculizumab group (21 of 43) had levels of hemoglobin that remained above the prespecified set point (the level at which each individual patient was transfused before starting trial medication; median 7.7 g/dl for both groups) in the absence of transfusions, whereas stabilization of hemoglobin levels did not occur in any patient in the placebo group (P < 0.001). By week 26, the median number of units of packed red cells transfused per patient was 0 in the eculizumab group and 10 in the placebo group (P < 0.001). In the 6-month period before the study, the median number of units of packed red cells transfused per patient was 9.0 in the eculizumab cohort and 8.5 in the placebo cohort. Transfusion independence was achieved in 51% of patients in the eculizumab group (22 of 43) and 0% of those in the placebo group (0 of 44, P <0.001). The overall mean rate of transfusion was reduced by 73% in the eculizumab group. Even among patients receiving eculizumab in whom transfusion independence was not reached, the number of units of packed red cells transfused was reduced by 44%, as compared with patients in the placebo group (P < 0.001). The median time to the first transfusion was 4 weeks in the placebo group and was not reached in the eculizumab group. Before treatment with eculizumab, the hemoglobin levels were maintained by transfusion. With eculizumab treatment, anemia was improved as indicated by the increase in endogenous erythrocyte mass, and the stabilization of hemoglobin levels with a concomitant cessation of or reduction in the number of transfusions.
A further phase III study, SHEPHERD (Safety and Efficacy of the Terminal Complement Inhibitor Eculizumab in Patients with Paroxysmal Nocturnal Hemoglobinuria), was an open-label, non-placebo controlled study designed to test eculizumab in a broader PNH population. 55 Patients only required one transfusion in the previous 24 months and platelet counts as low as 30 × 109 /L were allowed for study entry. Ninety-seven patients at 33 international sites were enrolled. The results supported those found in previous studies. Patients treated with eculizumab responded with an 87% reduction in hemolysis, as measured by LDH levels (P < 0.001). Baseline fatigue scores in the FACIT-Fatigue instrument improved by 12.2 +/− 1.1 points (P < 0.001). Eculizumab treatment led to an improvement in anemia. The increase in hemoglobin occurred despite a reduction in transfusion requirements from a median of 8.0 units packed red cells per patient pre-treatment to 0.0 units per patient during the study (P < 0.001). Overall, transfusions were reduced 52% from a mean of 12.3 to 5.9 units per patient. Forty-nine (51%) patients achieved transfusion independence for the entire 52 week period. Improvements in hemolysis, fatigue, and transfusion requirements with eculizumab were independent of baseline levels of hemolysis and degree of thrombocytopenia.
These 3 studies have demonstrated the significant benefit of eculizumab in a broad population of hemolytic PNH patients with varying preceding transfusion requirements. Eculizumab appears to have no effect on the underlying bone marrow hypoplasia and is therefore only indicated with evidence of hemolytic PNH. Although transfusions were significantly reduced and quality of life significantly improved, a proportion of patients still require transfusions and hemoglobin levels do not return to normal. Possible reasons include the underlying bone marrow failure or recognition of deposition of complement protein C3 on PNH red cells which may result in residual hemolysis. 56 The latter phenomenon has been recognized with the use of eculizumab, as prior to blockage of terminal complement, these cells would most likely have been lysed intravascularly.
Safety
No patient was withdrawn from eculizumab therapy due to an adverse event. Common adverse events are headache, nasopharyngitis and upper respiratory tract infection. No clinically significant anti-eculizumab antibodies have developed in any treated PNH patients. Two were reported in SHEPHERD with no disruption of complement inhibition. In the TRIUMPH study, one placebo-treated patient and one eculizumab-treated patient were reported to have a transient, low-titre antibody response to eculizumab. The major concern, as described above, is that of increased risk of Neisseria Meningitidis. During the trials, 2 of the 195 patients had meningococcal sepsis but made full recoveries with no sequelae following prompt therapy. Eculizumab therapy continues in both patients. Vaccination is required prior to therapy and we also recommend each treated patient carries an alert card.
Thrombosis
Preventing thrombosis in PNH is an important aim in the management of patients with PNH and would lead to reduced morbidity and mortality in this disease. Knowing that the tendency toward thrombosis in patients with PNH may involve high levels of free plasma hemoglobin with the consequent scavenging of nitric oxide (NO),26,57–61 exposure of prothrombotic erythrocyte membranes, and/or the absence of GPI-anchored complement inhibitors on the surfaces of circulating platelets, it is reasonable to hypothesize that thrombosis resulting from any or all of these mechanisms should be reduced by terminal complement blockade with eculizumab. In the pilot and extension studies using eculizumab in PNH no thrombotic events occurred during eculizumab treatment, although 6 of the 11 patients were on warfarin therapy before and during the study.12,50
The issue of possible protection against the risk of thrombosis through terminal complement inhibition with eculizumab was evaluated in the clinical studies of PNH. The prespecified clinical outcome of thromboembolism on an intention-to-treat basis in a multinational phase III open-label extension study that enrolled patients from 3 independent eculizumab PNH clinical studies (n = 195) was evaluated. These 3 studies were the phase II pilot study and its extensions,12,50 the phase III TRIUMPH study, 54 and the phase III SHEPHERD study55,62 and the common phase III extension study. For each study, the protocol specified that the principal investigator record the description, location, method of diagnosis, date of diagnosis, and date resolved for each thrombosis event in pre-identified case report form fields for events both prior to and during eculizumab treatment.
The pre-specified overall thromboembolism event rate was reduced from 7.37 events per 100 patient-years (124 total events) before eculizumab treatment to 1.07 events per 100 patient-years (3 total events) with eculizumab treatment in the same patients (P < 0.001). This represented a relative reduction of 85% and an absolute reduction of 6.3 thromboembolism events per 100 patient-years. The observed reduction of thromboembolism events during eculizumab treatment is unlikely to be due to improvements in patient care or more aggressive antithrombotic therapy leading up to and during the clinical studies since the thromboembolism event rate did not decrease in the period of time immediately preceding study enrolment or during the study in placebo-treated patients. The pre-eculizumab thromboembolism event rate during the 12-month period just prior to study initiation was markedly elevated, reaching a rate of 17.21 events per 100 patient-years.
Subclinical Thrombosis
A study using sensitive imaging techniques was employed to look for subclinical thromboses. 63 The results were very interesting as they have suggested an increased rate of thrombosis than previously reported. Ten PNH patients (median age 31.5 yrs) with large PNH clones but without previous clinical evidence of venous or arterial thrombosis and receiving supportive treatment underwent imaging. Five (50%) were receiving transfusions. Five (50%) of the patients were on primary anticoagulant prophylaxis with warfarin. Abnormalities suggestive of previous subclinical pulmonary thromboses were identified in 6 of 10 patients with PNH by high-resolution MR imaging, even in patients with recent disease onset. This is even more significant as this includes patients on primary prophylaxis with warfarin. As well as probable pulmonary thromboembolisms, there was evidence of subclinical myocardial damage in 2 of
10 patients and compromized cardiac function in the majority of patients. There were 5 patients in this cohort who were not transfusion dependent, yet they also had evidence of cardiac, renal and/or pulmonary pathology. There was evidence of renal pathology in 9 of 10 patients. No such subclinical thromboses would be anticipated in an age-matched control population. The finding of this high frequency of subclinical thromboses was surprising and reinforces the impact this disease can have in a relatively young group of patients.
A recent study has found that eculizumab treatment of patients with PNH results in a rapid decrease in plasma tissue factor microparticles, thrombin generation and inflammation as measured by D-Dimers, thrombin-antithrombin complexes and interleukin-6, 64 suggesting a further possible positive impact of eculizumab treatment in suppressing inflammation and prothrombotic activity in patients with PNH.
Possible Future Indications for Eculizumab
PNH is a fascinating disease and although many advances in the understanding of the pathogenesis of this disorder have been made over the last few decades, there are still areas that continue to be discovered and keep the disease at the cutting edge of research. The introduction of an effective therapy for a disease that is disabling in many patients has been at the center of many recent research studies.
Pulmonary Hypertension
Pulmonary arterial hypertension is an increasingly recognized complication of chronic hereditary and acquired hemolytic anemias and has been found to occur in approximately 30% of adult patients with sickle cell disease. 65 The finding of PHT has now been demonstrated in approximately 40% of patients with PNH and is believed to be linked to intravascular hemolysis. 66 Eculizumab has been shown to reduce NO consumption as well as prevent thrombosis through its amelioration of intravascular hemolysis.50,67 The incidence of PHT as measured by levels of B-type natriuretic peptide was found to be significantly reduced following eculizumab therapy in the phase III placebo-controlled trial, TRIUMPH. 68 This is therefore an area to explore as a potential indicator for therapy with eculizumab in the future.
Renal Impairment
Renal damage in PNH has been associated with chronic hemolysis and subsequent hemosiderosis and/or microvascular thrombosis. Renal impairment has been found to be common in hemolytic PNH. 69 All patients entering into the completed eculizumab multinational studies in hemolytic PNH patients were screened for chronic renal insufficiency (CRI) and for a previous clinical diagnosis of CRI and acute renal failure (ARF). The incidence of CRI at screening was 21% (40/195) with 10/40 patients having severe CRI (GFR ≤ 30 ml/min). Only 25% (10/40) of patients with pre-study CRI had been clinically diagnosed with CRI indicating the under-recognition of renal impairment in patients with PNH amongst hematologists. For patients with baseline CRI, GFR remained stable (median increase 0.47 ml/min/1.73m2) and 10% of patients were no longer classified as CRI with eculizumab treatment. Eculizumab was safe and effectively reduced hemolysis and thrombosis in PNH patients with ARF and CRI.
Pregnancy
During pregnancy, PNH frequently worsens, posing additional risk of morbidity and mortality to mother and fetus. Pregnancy was an exclusion criterion in the clinical trials using eculizumab in PNH. A recent abstract reported data on 6 pregnancies in patients receiving eculizumab. 70 There were no congenital anomaly/birth defects and no thromboses reported. Eculizumab appeared safe and well tolerated during pregnancy in these patients. A further report supports this conclusion. 71 Although small numbers of patients, these findings are very encouraging in a disease area where patients were previously discouraged from becoming pregnant.
Cold Agglutinin Disease (CAD)
Roth et al have reported on the chronic use of eculizumab in a patient with CAD refractory to previous treatments and experiencing elevated hemolysis for at least 1 year prior to treatment. 72 Based on the pathogenesis of CAD, eculizumab was used in this patient and hemolysis was significantly reduced and resulted in an improvement in anemia as well as reduced transfusion requirements. There were no further exacerbations of the disease.
Atypical Hemolytic Uremic Syndrome
Atypical hemolytic uraemic syndrome (aHUS) is a potentially life-threatening condition with most cases leading to end-stage renal failure. 73 Disruption of the complement system is also thought to play a role with mutations in genes encoding complement proteins being responsible in approximately 50% of cases. 74 Case reports have been published demonstrating the effective use of eculizumab in patients with aHUS.75,76 Both cases demonstrate the need to further evaluate eculizumab in this disease area.
Conclusion
Eculizumab effectively blocks terminal complement activation and has shown significant improvements in patients with PNH in terms of hemoglobin levels, transfusion requirements, thrombosis rate, quality of life, renal function, pulmonary pressure measurements and potentially pregnancy outcomes. The future for this drug includes the continuing treatment of hemolytic PNH patients, the need to explore other indications in PNH as well as other disease areas such as cold agglutinin disease and atypical hemolytic uremic syndrome.
Disclosure
AH has received honoraria from Alexion Pharmaceuticals, Inc. for lectures given.