
Abstract
Inotropic agents are often used to improve the contractile performance of the failing myocardium, but this is often at a cost of increased myocardial ischemia and arrhythmia. Myocyte contractility depends on the release of Ca2+ from the sarcoplasmic reticulum, and this Ca2+ is subject to regulation by the phosphorylation status of phospholamban (PLN). Many currently used inotropic agents function by increasing the phosphorylation of PLN, but these also heighten the risk of ischemia. Another approach is to reduce the dephosphorylation of PLN, which can be achieved by inhibiting pathways upstream or downstream of the protein kinase Cα. Phospholipase Cβ1b is responsible for activating protein kinase Cα, and its activity is substantially heightened in failing myocardium. We propose phospholipase Cβ1b, a cardiac-specific enzyme, as a promising target for the development of a new class of inotropic agents. By reversing changes that accompany the transition to heart failure, it may be possible to provide well-tolerated improvement in pump performance.
Introduction
Heart failure (HF) is a progressive, essentially irreversible disease in which the capacity of the heart to provide adequate blood supply is compromised. Along with its associated pathologies, heart failure contributes substantially to the cost of healthcare and the cost worldwide is rising rapidly. 1 The failing myocardium exhibits a range of pathologies, all of which need to be therapeutically targeted for the successful management of the condition. Treatment regimens commonly include angiotensin converting enzyme (ACE) inhibitors and β-adrenergic receptor blockers, and may incorporate an inotropic agent to enhance end organ perfusion.2,3 In patients with impaired systolic function, inotropic agents provide an improvement in cardiac performance, but this is often at a cost of increased myocardial oxygen consumption, potentially increasing the likelihood of ischemia and arrhythmia. 4 New types of inotropic agents are clearly needed. The failing myocardium exhibits a number of changes that contribute to contractile dysfunction. Included among these are loss of functional myocytes 5 and extracellular matrix deposition 6 ; but in addition to these structural changes, there are signaling changes within the myocytes themselves, which contribute to contractile dysfunction. These intracellular changes provide potential start points for developing novel therapeutic strategies. The overarching idea is that, by reversing the intracellular changes that accompany the transition to HF, it may be possible to provide well-tolerated improvements in the pump performance. In general, these would be used in combination with agents that target other aspects of the complex disease of HF.
Sarcoplasmic Reticulum Ca2+ as a Target for Inotropic Therapy
The regulation of cardiac contractile function is orchestrated primarily by the sarcoplasmic reticulum (SR), which provides the Ca2+ ions to initiate and sustain contraction of the myocyte. The cardiac contraction cycle is initiated by Ca2+ entry via voltage-regulated Ca2+ channels in the sarcolemma, which provide the trigger Ca2+ to activate ryanodine receptors on the SR, resulting in release of sufficient Ca2+ from the SR into the cytosol to initiate contraction during systole. 7 Ca2+ is subsequently pumped back into the SR by the sarco-endoplasmic reticulum ATPase (SERCA)2a, the activity of which is closely regulated by the phosphorylation status of phospholamban (PLN). PLN is an inhibitor of SERCA2a in the dephosphorylated state, and this inhibition is relieved by phosphorylation. 8 PLN is phosphorylated, most importantly, by protein kinase A (PKA) on S 16 following activation of β-adrenergic receptors, and its phosphorylation results in heightened SERCA function, accelerated relaxation, and increased SR Ca2+ content.
The Ca2+ content of the SR is typically lowered in failing myocytes, 9 and this is associated with reduced expression of SERCA2a, 10 along with changes in the expression or activity of upstream factors that regulate the phosphorylation status of PLN. PLN is typically dephosphorylated at S 16 in HF, 11 contributing to further lowered SERCA activity (Fig. 1). In addition to the positive regulation by PKA, S 16 phosphorylation of PLN is negatively regulated by protein kinase Cα (PKCα) 12 downstream of phospholipase Cβ1b (PLCβ1b). The expression and activity of both PKCα 13 and PLCβ1b 14 have been shown to be heightened in HF. Thus all of these factors are appropriate start points for the development new therapeutics.
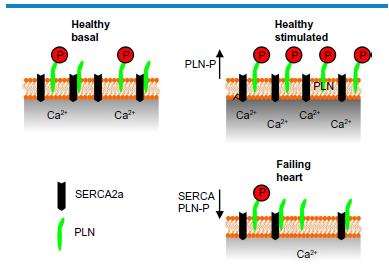
Diagram showing the changes in S 16 phosphorylation status of PLN in healthy heart under basal conditions, stimulated conditions, and in heart failure. A positive inotropic stimulus increases phosphorylation and dysinhibits SERCA2a. In failing cardiomyocytes, SERCA2a expression is depressed and PLN is dephosphorylated on S 16 . This results in lowered SR Ca2+ content.
SERCA2A
The expression of SERCA2a both at the mRNA and protein levels is depressed in most HF models independently of the etiology (Fig. 1). 11 , 15 Similarly lowered SERCA2a expression is a common feature of human HF 11 For this reason, SERCA2a has been a focal point for the development of an improved range of inotropic drugs. Pharmaceutical agents, such as the Na+/K+ ATPase inhibitor istaroxime, that increase SERCA activity have been tested with some positive outcomes in experimental and clinical studies, 16 but such agents have other actions that may present problems. Another approach is to reverse the lowered SERCA expression level directly by gene therapy, and considerable effort has been made in this regard. Virally mediated expression of SERCA2a in cardiomyocytes increased the amplitude of the Ca2+ transients, accelerated relaxation kinetics, and reduced diastolic Ca2+. 17 Subsequent studies used adenoviral constructs to deliver SERCA2a to hearts of rats that had been subjected to pressure overload. Restoration of the SERCA2a levels resulted in improved contractile performance along with substantially reduced mortality 18 Adenovirus presents difficulties in delivery to hearts in vivo and also instigates an immune response. For these reasons, recombinant adeno-associated viruses (rAAVs) have become the tool of choice for gene therapy to the myocardium, and delivery of SERCA2a has been foremost in these studies. The advantages of rAAV and the relative benefits of the different serotypes are discussed in detail elsewhere. 19 The development of rAAV vectors led to their use in large animal models of pacing-induced HF in sheep, 20 , 21 where delivery of the SERCA2a gene resulted in substantial improvements in contractile function along with decreased mortality. Significantly, translation into clinical studies has been initiated with encouraging results from completed Phase I and II clinical trials [calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID) 22 and CUPID2]. 23 These studies not only provided data supporting the usefulness of rAAV as a clinical tool but also showed that increasing SERCA2a had beneficial outcomes in the clinical situation.
Phospholamban Expression
In addition to its expression level, SERCA2a activity is dependent on the expression level and phosphorylation status of PLN (Fig. 1). However, attempts to improve contractile performance by manipulating PLN expression have generally met with less success than studies targeting SERCA2a. As expected, knockout of the PLN gene results in increased SR Ca2+ content and improved Ca2+ and contractile responses. Accordingly, knockout of PLN improved outcomes in a mouse model where calsequestrin was overexpressed, depressing available SR Ca2+. 24 However, in mice overexpressing CaMKIIδc in heart and displaying features of SR Ca2+ leak and associated arrhythmogenesis, deletion of PLN severely worsened phenotype by increasing Ca2+ leak and facilitating cardiomyocyte death following mitochondrial Ca2+ overload. 25 Given that heightened CaMKIIδ expression and activity are common features of failing myocardium, 26 removing PLN does not appear to be a useful approach. It is possible that more modest lowering of PLN expression levels might have provided better protection.
Phospholamban Phosphorylation
As noted above, PLN is an inhibitor of SERCA in the dephosphorylated state, and therefore increasing the phosphorylation of PLN at S 16 dysinhibits SERCA, increases SR Ca2+ content, and improves contractility 8 PLN is phosphorylated at S 16 primarily by PKA downstream of β-adrenergic receptor activation. 27 PLN is dephosphorylated principally by protein phosphatase-1 (PP-1), and PKA further regulates the phosphorylation status of PLN by phosphorylating a PP-1 inhibitor known as inhibitor-1 (I-1) at T 35 , thereby increasing its inhibitory function (Fig. 2). A number of currently used inotropic drugs act by increasing cAMP levels and thereby activating PKA, leading to increased PLN S 16 phosphorylation by these direct and indirect mechanisms. Included among these are β-adrenergic agonists, such as dobutamine, and cAMP phosphodiesterase inhibitors such as milrinone. 4 The downside of this approach is that PKA has other targets, some of which increase the energy demands on the heart and thereby promote ischemia, 28 a very undesirable side effect. For this reason, other approaches to increase PLN phosphorylation may be preferable.
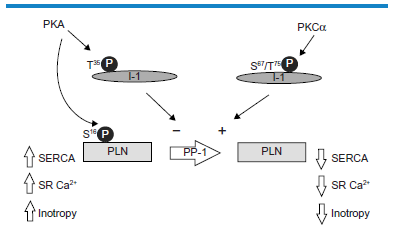
The regulation of S 16 phosphorylation of PLN downstream of PKA and PKCα. In addition to direct phosphorylation of PLN at S 16 , PKA phosphorylates I-1 at T 35 , depressing the activity of PP-1 and maintaining S 16 phosphorylation of PLN. PKCα phosphorylates I-1 at S 67 and T75, resulting in increased activity of PP-1 and dephosphorylated PLN with subsequent lowered contractile function.
PKCα
An alternative approach to improve contractility by increasing the phosphorylation of PLN at S 16 is to suppress inhibitory pathways. To date, this has been achieved by inhibiting PKCα. PKCα is a conventional PKC subtype, meaning that it requires both Ca2+ and sn-1,2-diacylglycerol (DAG) for activation, and it is the most highly expressed PKC subtype in rodent heart. 29 Cardiac PKCα activation results in contractile dysfunction associated with dephosphorylation of PLN and SR Ca2+ depletion. 12 The mechanisms of this response have been elucidated and are shown in Figure 2. Essentially, PKCα phosphorylation of I-1 at S 67 and or T75 opposes the action of PKA phosphorylation at T 35 . 30 Whereas PKA phosphorylation increases the inhibition of PP1 by I-1, PKCα phosphorylation of I-1 increases PP-1 activity, resulting in PLN dephosphorylation. PKCα thus acts to oppose β-adrenergic responses at the level of PLN. The obvious advantage of PKCα as a target over PKA is that PKCα activity is being inhibited rather than activated, and unwanted effects on energy metabolism are unlikely. Based on these considerations, PKCα inhibitors have been studied with a view to developing new inotropic therapies. 31
Overall, PKCα has been targeted by two different approaches. First, inhibitors of conventional PKC subtypes, with some specificity for PKCα, have been used with considerable success. 13 , 31 , 32 PKCα inhibitors prevented the loss of contractility in mouse hearts following pressure overload and increased survival, without altering the hypertrophic response. 33 Importantly, chronic treatment with the PKCαinhibitor ruboxistaurin improved survival in pigs following myocardial infarction (MI). 31 Ruboxistaurin is currently under development as a treatment for diabetic retinopathy. The other approach to targeting PKCα pathways to improve contractile function has involved the development of a mini-gene activator of PP-1 based on I–-1. This construct comprises the N-terminal sequence of I-1 (1–65, I-1c), lacking the PKCα phosphorylation sites and thus unable to function as a PP-1 activator. Instead, this construct inhibits PP-1 unopposed and thereby increases SERCA activity, SR Ca2+ content, and contractile function. Studies in a post-MI model in pigs provided evidence for substantial improvement in functional parameters and survival following rAAV9-mediated expression of I-1c. 34 However, other investigations using a mouse model where I-1c was expressed in hearts of I-1–/– mice showed initial improvements, but with aging the mice developed a cardiomyopathic phenotype associated with hyperphosphorylation of PLN and the ryanodine receptor, Ca2+ sparks, and ventricular tachycardia. 35 There were two major differences between these studies that might explain the divergent outcomes. First, the studies using the mouse model were carried out over a much longer time frame than the studies of the post-MI model in pigs, suggesting that chronic treatment with I-1c might be deleterious. Second, the chronic expression of I-1c in the mouse study was on a background of I-1 knockout, and this would be expected to intensify the effect of the PP-1 activator. This suggests that dosage might be critical for successful use of this mini-gene strategy for long-term therapy. A more recent study delivered I-1c to pigs via a modified rAAV vector and reported considerable improvement in cardiac function post MI, laying the basis for potential gene therapy. 36
In addition to the two mechanisms described above, recent evidence suggests that PKCα can be inhibited by overexpressing PICOT (PKC-interacting cousin of thioredoxin). PICOT has anti-hypertrophic actions and improves contractile function. Recent studies report that the positive inotropic action of PICOT depends on its ability to inhibit PKCζ, resulting in reduced expression of PKCα, increased PLN phosphorylation, and heightened SR Ca2+ content.37,38
PLCβ1b-Shank3
PKCα is expressed and is active in all cell types, 39 and therefore the use of PKCα inhibitors may be constrained by unwanted actions in other tissues. Another approach to maintaining the phosphorylation status of PLN is by preventing the activation of PKCα. PKCα is a conventional PKC subtype and therefore requires Ca2+ and DAG for activation. 40 DAG is generated by PLC enzymes following activation of appropriate receptors, 41 and therefore a PLC subtype or subtypes must be the immediate upstream activator of PKCα.
PLC enzymes hydrolyze the plasma membrane phospholipid phosphatidylinositol(4,5)bisphosphate (PIP2) to generate inositol(1,4,5)trisphosphate (IP3) and DAG. 42 IP3 is a regulator of Ca2+, 43 and, as noted above, DAG is an activator of conventional and novel PKC family members. 39 Cardiomyocytes express a number of different PLC subtypes, specifically PLCβ1 (of which there are two splice variants PLCβ1a and PLCβ1b), PLCβ3, PLCγ1, PLCδ1, and PLCε.14,44 The PLCβ family members are activated by Gq and thus by G protein coupled receptors (GPCR) including α1-adrenergic receptors, angiotensin II receptors (AT1), and endothelin receptors.45–47 In addition to activation by Gαq, PLCβ subtypes require translocation to the plasma membrane for activity, and for most PLCβ subtypes this is achieved by binding of a C-terminal PDZ-interacting domain to the PDZ domain of a particular protein scaffold. PLCγ is activated by plasma membrane translocation as part of the signaling cascade of many growth factor receptors, and this is associated with tyrosine phosphorylation and Src homology 2 (SH2) interactions. 42 PLCδ subtypes associate with the plasma membrane via a high-affinity PH domain specific for PIP2. They do not respond to regulatory factors other than Ca2+, and their functional significance remains poorly documented. 48 PLCepsiv; is a multifunctional protein with both PLC and GEF (guanyl nucleotide exchange factor) activities. 49 Activation of PLCε is complex, involving primarily monomeric G proteins of the Rho and Ras families, and can be initiated by receptors coupled to G12/13. Additionally, PLCε can be activated downstream of receptors coupled to Gs and adenylyl cyclase following cAMP activation of EPAC (exchange protein activated by cAMP), which generates activated Rap. 50 The relationship between the different classes of PLC expressed in cardiomyocytes is depicted in Figure 3.
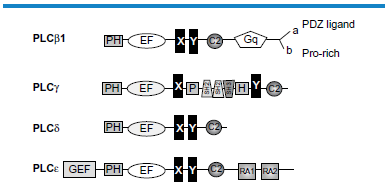
The subtypes of PLC expressed in cardiomyocytes. All PLCs share the X and Y domains that form the active site. This is separated by a linker region that functions as an inhibitor. In PLCγ, the X and Y domains, as well as a PH domain, are separated by SH2 and SH3 domains. PH, plextrin homology domain, a domain that binds PIP2; EF, EF hand domains that bind Ca2+; C2, C2 domain, a Ca2+ binding motif associated with membrane association; RA1 and RA2, Ras interacting domains; Gq, the Gq interaction site involved in activation of PLCβ subtypes. GEF, CDC25 GEF guanine nucleotide exchange factor domain.
PLCβ subtypes are generally believed to be the primary effectors of Gq activation in heart, 51 and our studies have shown that Gq responses in cardiomyocytes are mediated solely by an unusual member of the PLCβ family, PLCβ1b. 52 There have been suggestions of a role for PLC in cardiomyocyte responses to IGF1 (insulin-like growth factor-1), 53 and this would most likely be PLCγ, although this was not verified. In our hands, overexpression of PLCγ1 in cardiomyocytes did not alter responses to either EGF or PDGF. 54 Overexression of PLCδ1 in cardiomyocytes increased PLC activity but did not alter cardiomyocyte morphology in any obvious way.54,55 Furthermore, knockdown of PLCδ1 did not alter overall PLC activity in cardiomyocytes, even under conditions of heightened Ca2+. 56 The roles of PLCε in heart are complex. Both anti-hypertrophic and pro-hypertrophic actions have been reported, depending on the developmental stage of the heart. 44 , 57 , 58 In addition to a role in hypertrophic responses, PLCε facilitates contractile responses downstream of β-adrenergic receptor activation by enhancing systolic Ca2+ responses by mechanisms involving CaMKIIδ and PKCε. 57
Thus, PKCε is associated with pathways downstream of PLCε that increase cardiomyocyte Ca2+ responses and contractile activity. As noted above, PKCα, in marked contrast, reduces Ca2+ responses and lowers contractile performance. Our studies showed that, in the heart, PKCα was activated specifically by PLCβ1b. PLCβ1b expression and activity are selectively heightened in diseased myocardium from humans, sheep, and mice, and, furthermore, PLC activity correlates with disease progression, hinting at a role in the disease process. 14 This view was subsequently confirmed. Increasing the expression of PLCβ1b in mouse hearts resulted in a rapid loss of contractile function. In the continued presence of high PLCβ1b activity, contractile dysfunction was sustained for a period of 36 weeks, without indications of HF 59 PLCβ1b expression resulted in lowered S 16 phosphorylation of PLN and depressed SR Ca2+ content. All of these responses closely resemble changes that follow heightened PKCα expression. 12 Importantly, inhibition of PKCα resulted in complete restoration of contractile activity in PLCβ1b-expressing mice, confirming that PLCβ1b is the upstream activator of PKCα in vivo.
PLCβ1b is therefore a suitable target for the development of new inotropic drugs. However, direct inhibitors of PLC catalytic activity are unlikely to be successful. As described above, the active sites of the various PLC subtypes share considerable homology, and developing an inhibitor with subtype specificity would be challenging. 42 PLCε has positive inotropic actions, and therefore a general inhibitor of PLC catalytic activity might cause unwanted cardiac responses. In any case, there are currently no credible PLC inhibitors available as start compounds. U-73122 is often used as PLC inhibitor, but it has multiple actions and has never been shown to directly inhibit PLC.60–65
There is, however, a way in which PLCβ1b can be inhibited in cardiomyocytes in a selective manner. PLCβ1b is an atypical splice variant of PLCβ1, and it differs from all other PLCβ subtypes in having a C-terminal proline-rich sequence instead of the usual PDZ-interacting domain (Fig. 3). 42 , 66 , 67 In general, PLCβ subtypes target their substrate PIP2 by interacting with a PDZ protein scaffold. 67 The exchange of the PDZ ligand for a proline-rich sequence suggests that PLCβ1b is activated by a unique mechanism. We subsequently identified the SH3 domain and ankyrin repeat protein 3 (Shank3) as the protein scaffold required for the localization of PLCβ1b at the cardiac sarcolemma, where it is active. 68 Shank3 is a high molecular weight multidomain protein that incorporates an SH3 domain in addition to a PDZ domain, a proline-rich sequence, and ankyrin repeats. 69 The SH3 domain is the attachment point for the proline-rich sequence at the C-terminal end of PLCβ1b. Like PLCβ1b, Shank3 is expressed in only a limited number of cell types, 69 , 70 and so the PLCβ1b–Shank3 interface represents a cardiac-specific signaling system. This has been verified in studies showing that expressing the splice-variant-specific C-terminal sequence of PLCβ1b (PLCβ1b-CT), as a mini-gene, disrupted the interaction between PLCβ1b and Shank3 and prevented downstream signaling. 52 More recent studies have confirmed that delivery of PLCβ1b-CT to mouse heart in vivo protected it from contractile dysfunction following pressure overload, confirming the usefulness of this interface as a start point for the development of inotropic agents (Fig. 4).
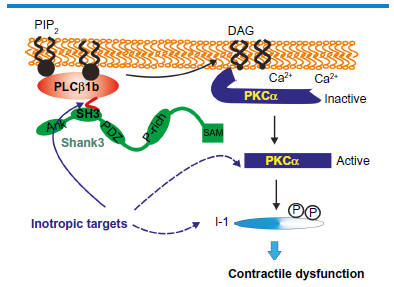
Signaling pathways downstream of PLCβ1b and how they can be targeted for drug development. PLCβ1b generates DAG and activates PKCα leading to I-1 phosphorylation and contractile depression. PLCβ1b requires its C-terminal interaction with Shank3 for activity, providing a cardiac-specific interface. Ank, ankyrin-repeat sequence; SH3, SH3 domain; PDZ, PDZ domain; P-rich, proline-rich sequence; SAM, sterile alpha motif. The blue arrows indicate current (dashed lines) or potential (solid line) targets for the development of inotropic drugs.
Summary and Conclusions
Maintaining the phosphorylation status of PLN as a means to optimize SERCA2a activity and maintain SR Ca2+ content is a well-substantiated approach to inotropic therapy. There is a substantial body of work that validates inhibiting the inhibitory pathway mediated by PKCα as a way to achieve this aim. As discussed, PKCα itself is not a cardiac-specific target, and focusing on the upstream activator PLCβ1b may provide a better option. The interaction between the proline-rich sequences in the C-terminal tail of PLCβ1b and the SH3 domain of Shank3 provides a target that is readily amenable to high-throughput compound screening or to drug design based on structural analysis.
Author Contributions
Wrote the first draft of the manuscript: EAW. Contributed to the writing of the manuscript: DRG. Agree with manuscript results and conclusions: EAW, DRG. Made critical revisions and approved final version: DRG. Both authors reviewed and approved of the final manuscript.