
Abstract
Darunavir is an oral peptidomimetic HIV-1 protease inhibitor with antiretroviral activity against wild type HIV strains and HIV strains with protease inhibitor mutations. Ritonavir-boosted darunavir is rapidly absorbed and it has a higher bioavailability than unboosted darunavir. In HIV infected adults, the pharmacokinetic profile of darunavir showed that the drug concentrations are similar in the age range between 18 to 65 years and unaffected by the presence of moderate renal or hepatic function impairment. Darunavir chemical structure provides a strong interaction between the drug and HIV-1 protease and accounts for its potent antiretroviral activity and high genetic barrier to the development of resistance. The efficacy, safety and tolerability of darunavir have been widely demonstrated in HIV-infected treatment-experienced and naïve adult patients and it's use has been labelled in this population. Recently, upon the approval of the Food and Drug Administration, a 75 mg darunavir's tablet formulation has been licensed for the treatment of HIV-infected children and adolescents in the age range between 6 to 17 years.
Introduction
Effective antiretroviral combination therapy dramatically reduced HIV-related morbidity and mortality,1,2 but the development of viral drug resistance, which occurs as a result of mutations in the viral genome,3,4 continues to be responsible of treatment failure.
Darunavir is a protease inhibitor (PI) recently approved in several countries of the European Union and in the United States. Ritonavir-boosted darunavir (DRV/r) demonstrated an in vitro substantial antiretroviral activity against wild-type HIV strains and HIV strains with protease inhibitors (PIs) mutations, and sustained efficacy and safety in naïve and treatment-experienced HIV-infected patients.5–8 This review describes the pharmacokinetic and pharmacodynamic profile, pharmacologic interactions of the drug and focuses on the clinical use in HIV-infected patients.
Pharmacokinetic Properties
Pharmacokinetic parameters of darunavir administered orally with low-dose ritonavir tablet or solution formulations have been recently assessed in different studies on healthy volunteers and treatment-experienced and naïve HIV-infected patients.9–14
Absorption and distribution
DRV/r orally administered has demonstrated a rapid absorption.11–16 Different dosages have been assessed and the recommended daily dose of darunavir in treatment-experienced adult patients is 600 mg with ritonavir 100 mg twice daily (bid), while in treatment-naive adult patients is 800 mg with ritonavir 100 mg once daily.15,16
Darunavir in vitro is mainly absorbed through passive transcellular diffusion. 17 The bioavailability of a 600 mg dose of darunavir alone is 37% and peak plasma concentrations are obtained within 2.5 and 4 hours. 16 Owing to an overall net inhibition of CYP3A4 and P-glycoprotein activity by ritonavir, the addition of 100 mg of ritonavir increases the bioavailability up to 82% and results in a 14-fold increase in the darunavir area under the plasma concentration-time curve (AUC) compared with administration of darunavir alone. 10 A further increase in bioavailability up to 30% is observed when the drug is taken with food and it is comparable for different types of meals assessed.18,19 The in vitro binding of Darunavir to plasma proteins ranges from 92% to 94%. 10 Darunavir is mainly bound to α-acid glycoprotein,20,21 while a minor amount of the drug is bound to albumin. 11 Raising darunavir concentrations results in unbound darunavir increase. 9 Darunavir concentrations were also assessed in 14 cerebrospinal fluids from eight HIV-1-infected individuals. 22 All samples had detectable drug concentrations with a median value of 34.2 ng/ml (range 15.9–212.0 ng/ml) and most of them were in the range of levels needed to inhibit replication of wild type virus. This property may account for darunavir contribution to the suppression of HIV replication in the central nervous system. 22
Metabolism and elimination
In vitro studies have identified oxidative process as a major pathway of darunavir metabolism. 23 Hepatic cytochrome P450 (CYP) enzymes are first implicated in drug metabolism, particularly CYP3A4. 23 As ritonavir acts as an inhibitor of CYP3A4 enzyme and P-glycoprotein enzyme activity, coadministration of darunavir plus 100 mg ritonavir reduces darunavir metabolism and increases its serum concentration by ≍14 fold versus darunavir alone. 10 The mean elimination half-life of DRV/r in a dose-ranging study on healthy volunteers was found to be 15 hour; most of darunavir (79,5%) was eliminated through faeces and to a lesser extent (13,9%) through urine. 24 Similarly, the mean terminal elimination half-life of darunavir (time required to divide the plasma concentration by two after reaching pseudo-equilibrium) with various dose regimens ranged 10.9–17.2 hours. 11
Darunavir pharmacokinetic profile in HIV adults shows no differences across the age range of 18 to 65 years. Darunavir exposure in subjects 6 to 18 years of age receiving weight based dosages matched that of treatment experienced adults receiving a DRV/r standard dose. 25 Pharmacokinetics parameters, safety, tolerability and efficacy of darunavir in patients ranging 3 to 6 years or older than 65 years of age have not been established yet.
Pharmacodynamic
Darunavir is a PI similar to amprenavir. 21 Darunavir chemical conformation contains a bis-tatrahydrofura-nylurethane (bis-THF) moiety and a sulphonamide isostere and darunavir is administered as its ethanolate salt. 8 The drug's chemical structure has two THF rings fused together, thus differing from Amprenavir which has one ring only. The bis-THF moiety conformation is also provided by at least six hydrogens that strengthen the interaction between darunavir and a key HIV-1 protease amino acid, the Asp 29. 8 Darunavir has shown high binding affinity and potent in vitro activity against HIV-1 wild type protease. 27 The drug largely binds the wild-type HIV-1 protease (Kd 3.9 x 10–10 mol/L), thus preventing viral protein cleavage of HIV-1 Gag-Pol poliproteins in infected cells and production of infectious viral particles. 26
Decreased binding affinity with multi drug resistant (MDR) HIV-1 protease has been shown for darunavir; nevertheless, stronger interaction, more than 33-times compared to amprenavir and more than 10-fold tighter compared to that of first generation PIs (nelfinavir or lopinavir) has been described between darunavir and MDR HIV-1 protease. 27
Darunavir in vitro antiretroviral activity has been described against wild type HIV-1, subtypes A to H, with a median 50% effective concentration (EC50) ranging from 1 to 5 nM and the corresponding median 90% effective concentration (EC90) ranging from 2.7 to 12 nM. Moreover, darunavir has exhibited an EC50 of 4.2 nM and a median EC90 of 13 nM against wild type HIV-2, and these are comparable with the activity observed against HIV-1. 20 Furthermore, darunavir has remained active against a panel of clinical isolates resistant to an average of 5 first generation PIs with an EC50 of <10 nM for all isolates. 20 Further studies defined the EC50 as the stronger predictor of darunavir efficacy in Power 1, 2, and 3 analysis.28,29 From a multivariate analysis of the data obtained from Power 1 and Power 2 clinical studies the inhibitory quotient, defined as the ratio between darunavir through steady state concentration and baseline EC50, resulted to be another factor strongly predicting the virological response and decrease in viral load at week 24. 9
Additional antiretroviral activity and no antagonism have been highlighted between darunavir and any of the nucleoside reverse transcriptase inhibitors (NRTIs) abacavir, didanosine, lamivudine, stavudine, zalcitabine, zidovudine, and tenofovir; nonnucleoside reverse transcriptase inhibitors (NNRTIs) delavirdine, efavirenz, and nevirapine; PIs indinavir, ritonavir, nelfinavir, saquinavir, amprenavir, lopinavir, and atazanavir or the fusion inhibitor T-20. Some evidence of synergy has been observed between darunavir and the PIs ritonavir, nelfinavir and amprenavir. 5
Impact of PI resistance mutations
The potent activity of DRV/r has been confirmed in patients failing a PI-containing regimen in Power 1, 2 and 3 studies,6,7 as in moderately pre-treated patients, 30 and in antiretroviral naive patients. 31
Multiple PI-resistant HIV-1 isolates have been collected form PI-experienced patients receiving darunavir 600/100 mg bid who experienced virological failure. 32 A series of 11 mutations, V11I, V32I, L33F, I47V, I50V, I54L/M, G73S, L76V, I84V, L89V associated with a reduced in-vitro susceptibility and virological response to darunavir have been identified. 32 Nevertheless, efficacy of treatment was higher in those receiving boosted darunavir than boosted control PIs. Superior efficacy of darunavir was confirmed regardless of the number of baseline mutations. 33 The manufacturer's prescribing information also describes association between the presence at baseline of two or more of the substitutions V32I, I54L/M, V11I, I15V, L33F, I47V, I50V, L89V and a decreased virologic response in patients receiving DRV/r. 34 Previous PIs or amprenavir/fosamprenavir therapies and have also been associated with a greater risk of developing darunavir resistant associated mutations. 35
Analysis of genotypes performed in 25 patients experiencing virological failure on a DRV/r containing regimen revealed darunavir associated resistance mutations in 72% of patients at codons L89I/M/V, V32I, V11I, I47V/A, I54L/M, L33F/I, and I50V. 36 Recent study has analyzed the impact of individual protease gene mutations on the virological response to DRV/r: eight mutations with a negative impact on the virological response (K14R, K20I, E34Q, I47V, I54M, K55R, T74P and I84V) and three mutations (K20R, E35D and V82A) with a positive impact were identified. 37 A genotypic score was created and this resulted highly predictive of the virological response at month 3, along with the baseline plasma viral load and the baseline CD4 cell count. No association with the virological response was observed with the previous use of any PIs. Two mutations, I437T/V in gag and L76V in protease have been recently associated with lower rate of selection of darunavir resistance mutations in 48 PI-experienced patients failing treatment on a DRV/r containing regimen. Mutations in the natural substrate rather than the enzyme itself may be a novel mechanism of HIV resistance to PIs. Viruses with the I437T/V mutation probably have increased protease efficiency due to mutated substrate and do not require darunavir resistance mutations in the protease gene to be resistant to darunavir. 37
Cross resistance to other PIs and darunavir has been observed; in particular, mutations correlated to a reduced clinical response to darunavir are similar to those known to confer reduced susceptibility to amprenavir. 38 However, a less than tenfold decreased susceptibility is reported for darunavir in cell culture against 90% of 3309 clinical isolates resistant to atazanavir and other first generation PIs; 34 this data explains why the viruses resistant to these PIs remain susceptible to darunavir. In darunavir-resistant viruses no susceptibility to amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, or saquinavir was observed in cell culture. Fourty-one of the viruses isolated from patients experiencing virological failure on darunavir standard dose remained sensitive to tripanavir, less than 10% remained susceptible to saquinavir and 2% to other PIs. Cross resistance between darunavir and NNRTIs, NRTIs, or fusion inhibitors, CCR5 co-receptor antagonists, or integrase inhibitors is unlikely because of different viral targets.39,40
Pharmacologic Interactions
DRV/r is an inhibitor and substrate of CYP3A and CYP2D6. Its coadministration with drugs that are primarily metabolized by CYP3A and CYP2D6 is expected to alter its plasma concentrations and therapeutic effect. Drugs that induce CYP3A activity would be expected to increase the clearance of darunavir and ritonavir and to lower their plasma concentrations. Here below are listed the most clinically relevant interactions.
Antibacterials
Significant decreases in DRV/r 600 mg/100 mg bid plasma concentrations and loss of therapeutic effect are described in combination with rifampicin, a potent inducer of CYP3A4 enzyme activity; coadministration is thus not recommended. 34 DRV/r may conversely increase the plasma concentrations of clarithromicine, by the inhibition of the isoenzyme responsible for the metabolic clearance of clarithromicine. As CYP3A4 also mediates the formation of the 14OH-clarithromycin, the primary and active metabolite of clarithromicine, this may result extensively inhibited. 41 However, no adjustment to the licensed doses of darunavir or clarithromycin is required for patients with normal renal function. 41 Caution is warranted for patients with renal impairment and a dose reduction of clarithromycin should be taken into consideration. 34 Coadministration of DRV/r and rifabutin is expected to increase rifabutin plasma concentrations; 42 a reduction of rifabutin dosage by at least 75% of the usual dosage, with a maximum dose of 150 mg once every other day should be considered. 42 Caution must also be taken when DRV/r is coadministered with metronidazol due to the alcohol contained in ritonavir formulation and the risk of disulfiram-like reactions. 15
Anticonvulsants
No clinical interaction is expected in the coadministration of DRV/r with gabapentin, lamotrigine, levetiracetam, valproate and vigabatrin. 34 The coadministration of DRV/r with carbamazepin 200 mg twice daily reduced ritonavir AUC by 49% and increased carbamazepine AUC by 45%. 34 No dose modification of DRV/r or carbamazepine is suggested; nevertheless, drug monitoring is required and carbamazepine dosage may need to be reduced from 25% to 50%. 34 Phenobarbital and phenytoin are inducers of CYP450 enzymes. According to the European medicines Agency, ritonavir boosted darunavir should not be used in combination with phenobarbital and phenytoin as coadministration may cause significant decreases in darunavir plasma concentrations. 16
Antidepressant
Paroxetine is mainly metabolised by CYP2D6, which plays a minor role in ritonavir metabolism. Low dose of DRV/r (400/100 mg bid) is associated with decreased paroxetine (20 mg once daily) AUC, peak plasma concentration (Cmax) and minimum plasma concentration (Cmin) by 39%, 36% and 37% respectively and no changes in darunavir exposure. 43 Titration of paroxetine dose must be considered according to clinical response. Caution must be used as a similar effect is expected with sertraline. 43 Nausea, dizziness, hypotension and syncope might be observed with trazodone or desipramine, due to the potential increase of their concentrations. Caution and lower dose must be considered. 34
Antifungals
No clinically relevant interaction is expected with the following antimycotic agents: amphotericin, fluconazole, flucytosine, terbinafine. Caution must be taken with caspofungin, itraconazole, miconazole, posaconazole, and voriconazole. Itraconazole and ketoconazole are potent inhibitors as well as substrates of CYP3A4. 34 Thus, their concentrations might increase in coadministration with DRV/r. According to the existing data, doses higher then 200 mg of ketokonazole should be avoided when combined with DRV/r. 44
Antihistamines
Co-administration of ritonavir is contraindicated with astemizole and terfenadine, 34 as these are highly dependent on CYP3A4 for clearance. 45 Prolongation of the QT interval and ventricular arrhythmias have been described. Potential interaction requires close monitoring; modification of drug dosage or timing of administration might be necessary when darunavir is combined with fexofenadine or loratadine. No contraindication is reported for cetirizine. 34
Antiplatelet and anticoagulant
Warfarin is primarily metabolised by CYP2C9. The coadministration with DRV/r and warfarin (10 mg single dose) resulted in a decrease of the Cmax and AUC of S-warfarin (active metabolite of warfarin) by 8% and 21% respectively. It is therefore recommended to monitor the international normalized ratio when warfarin is combined with DRV/r. 46
Antiretrovirals
Decreased darunavir AUC, Cmax and Cmin by a mean of 26%,17% and 42% respectively have been observed in coadministration with saquinavir 1000 mg. 47 A decrease in saquinavir exposure has also been reported (AUC12 and Cmax by 6% and the Cmin by 18%). Furthermore, a combination therapy with DRV/r plus saquinavir showed increased incidence of adverse events. 47 Coadministration of saquinavir and darunavir is thus contraindicated. A decline in darunavir exposure and its therapeutic effect have also been described in combination with lopinavir/ritonavir. Even though minimal influence has resulted on lopinavir exposure (~9% increase in the AUC12), 48 and no increases in adverse events have been reported with this combination of PIs, the coadministration of DRV/r and lopinavir is not recommended. 34 Appropriate dose of indinavir given in combination with DRV/r has not been yet established. Nevertheless, an increase of both indinavir (AUC and Cmin increase by 23% and 125%) and darunavir concentrations (AUC and Cmin increase by 24% and 44%) are reported combining twice daily administration of DRV/r 400/100 mg bid and indinavir 800 mg bid. 34 A reduction from 800 mg bid to 600 mg bid of indinavir is suggested in cases of intolerance to this combination. 34 No adjustment in dose is necessary when atazanavir is administered with DRV/r. 34
Maraviroc acts as a CYP3A4 substrate and DRV/r raises its concentrations by inhibiting its metabolism; increase of maraviroc AUC and Cmax 3 to 4 folds have been demonstrated in a combined therapy with DRV/r.49,50 DRV/r exposure did not result significantly altered. According to this data, the maraviroc dose should be decreased by 50% of the US Food and Drugs Administration (FDA)-approved dosage (from 300 mg to 150 mg bid). The effects of DRV/r on enfuvirtide have not been investigated. Increased AUC, Cmax and Cmin of T20 by 22%, 24% and 14%, respectively were demonstrated in coadministration with ritonavir alone 200 mg bid. 51 Nevertheless, results from in vitro and in vivo studies suggest that enfuvirtide is unlikely to have significant drug interactions with darunavir. 51 Nevirapine combined with darunavir showed decreased clearance and distribution volume and increased trough plasma concentrations; 52 however, no need for dose adjustment have been suggested when DRV/r 400 mg/100 mg is coadministered with nevirapine at the licenced dose of 200 mg bid. 53 No significant increases and no increased adverse reactions have been observed combining DRV/r 300 mg/100 mg and tenofovir 300 mg; thus, adjustment of tenofovir dose is not required. 54
Plasma concentrations of Etravirine at 100 mg bid decreased in coadministration with DRV/r at the standard dose, while etravirine 200 mg bid reaches higher concentrations. Thus, etravirine 200 mg bid can be administered with DRV/r without dose adjustment. 55 Caution should be taken when coadministration occurs with darunavir and efavirenz. 56 Since DRV/r inhibits CYP3A4 and efavirenz induces CYP3A4, and a decrease in darunavir exposure and an increase in efavirenz exposure can occur. 56 No dose adjustment is though required; however, clinical monitoring is indicated as possible central nervous system toxicity may be associated with the increased exposure to efavirenz. 34
Lipid lowering drugs
Atorvastatin is a common HMG-CoA reductase used in the management of hyperlipidemia; most of this drug is bound to plasma proteins and metabolized by CYP3A4 to its active metabolite. 57 Atorvastatin 10 mg coadministered with DRV/r 300 mg/100 mg have an AUC24 only 15% lower than the standard 40 mg administered alone, 58 while no modification in pharmacokinetics of DRV/r is induced by atorvastatin. A 10 mg once daily provides the correct atorvastatine exposure; a gradual dose increase of the compound may be tailored to the clinical response. 34 In 14 HIV subjects pravastatin 40 mg single dose coadministered with DRV/r showed a maximum increase in Cmax and AUC of 63% and 81% respectively; thus, it is recommended to start with the lowest possible dose of pravastatin and titrate as necessary. 59 Lovastatin and simvastatin highly depend on CYP3A4 metabolism, and their concentrations are expected to increase when coadministered with DRV/r. Myopathy, including rhabdomyolysis, has been reported as a side effect. Concomitant use of DRV/r with lovastatin and simvastatin is therefore contraindicated. 34
Oral Contraceptives
Efficacy and safety of ethinylestradiol/norethisterone 0.035/1.0 mg once daily decreases in coadministration with DRV/r standard dose; at the dosage of 0.035 mg, a mean ethinylestradiol decline of the AUC24, Cmax and Cmin by 44%, 32% and 62%, respectively have been observed. Similarly, the mean AUC 24, Cmax and Cmin of norethisterone 1.0 mg declined 14%,10% and 30% respectively when administered concurrently with DRV/r standard dose. No significant alteration in DRV/r concentrations were observed. 60 Alternative or additional contraceptives are therefore indicated. 34
Other drugs
Multiple studies have shown that both histamine H2 receptors such as ranitidine or proton pump inhibitors omeprazole combinations are well tolerated and had no significant effect on the pharmacokinetics of DRV/r. 61 Plasma concentrations of calcium channel blockers, including felodipine, nifedipine, and nicardipine, may increase when given concurrently with DRV/r. Caution is warranted and clinical monitoring of patients is recommended. 34 Plasma concentrations of immunosuppressants, including tacrolimus and sirolimus, may be increased when coadministered with DRV/r. Therapeutic concentration monitoring for the immunosuppressive agent is recommended when these drugs are taken concurrently. 34 When methadone is coadministered with DRV/r, patients should be monitored for abstinence syndrome, as ritonavir is known to induce the metabolism of methadone, leading to a decrease in methadone's concentrations. An increase in methadone dosage may be considered based on the clinical response. 62 Concomitant administration of DRV/r with PDE-5 inhibitors, including sildenafil, vardenafil, and tadalafil, should be applied with caution. PDE-5 inhibitor dosing should not exceed those indicated by the manufacturer. 34
Clinical Studies: Treatment Efficacy
Treatment-experienced adult patients
POWER (Performance of TMC114/r When Evaluated in treatment-Experienced patients with PI resistance) 1 and POWER 2 were randomized, multicentre, 144 weeks phase IIb studies that compared the efficacy and safety of darunavir plus low-dose ritonavir versus investigator-select control PIs. The first 24 week of POWER 1 and 2 were partially blinded dose-finding studies,63,64 followed by open-label phases at 48, 6 96, 65 144 weeks. 66 POWER 3 is an analysis of two Phase IIb, noncomparative, open-label, 144-week trials in patients treated with darunavir/r 600/100 mg bid.7,67 Inclusion criteria were: HIV infected-patients aged at least 18 years old with viral load >1000 copies/ml previously treated with highly active antiretroviral therapy (HAART defined as combination of 3 or more anti-viral drugs) for at least 3 months and having one or more PI mutations. Exclusion criteria were AIDS–-defining illness, acute liver disease, liver impairment function and use of any investigational antiretroviral drugs. Patients with chronic hepatitis B (HBV) or C (HCV) entered in POWER 1 trial as long as their condition was clinically stable but not in POWER 2 trial. Prior to randomization screening genotype profile was performed in all patients to choose an Optimized Background Regimen (OBR) consisting at least two NRTIs with or without enfurvitide. The use of NNRTIs was excluded. Screening for the number of primary PI mutation number was also performed. At randomization patients were assigned to boosted darunavir arm (400/100 once daily, 800/100 once daily, 400/100 bid or 600/100 bid) or to comparator boosted PIs arm. The most frequently used PIs were lopinavir, saquinavir, fosamprenavir and atazanavir. At week 24 all DRV/r patients switched to DRV/r 600-100 mg bid whereas patients receiving PIs continued with their assigned treatment. The treatment was discontinued in case of: virological failure defined as viral load reduction <0.5 log copies/ml at week 8; <1.0 log10 copies/ml at or beyond week 12 or an increase of 0.5 log copies/ml above nadir; tolerability and toxicity causes The primary efficacy end point was confirmed viral load reduction >1.0 log10 copies/ml or more from baseline (time-to-loss of virological response [TLOVR] algorithm).6,63–68 The secondary efficacy end points were the proportion of patients reaching viral loads less than 50 and 400 copies/ml (TLOVR), change in HIV-RNA and CD4 cell count from baseline. Safety and tolerability were also continuously assessed. The efficacy and safety analyses were based on the intent-to-treat (ITT) population which included all those who had received at least one dose of the study medication.6,7,63,64 The combined 46, 98, 144 analysis included only patients who received boosted DRV/r 600/100 mg bid or boosted PI from randomization subgroup.6,64,65 In POWER 1, 63 and 2, 64 after 24 weeks of treatment more patients treated with DRV/r achieved the primary end point than those treated with PI group. This result was confirmed at week 48, 6 96, 65 and 144. 66 Considering the secondary end points, the percentage of patients achieving a HIV-RNA < 50 copies/ml and the mean increase in CD4 cell count were higher in DRV/r arm particulary in patients treated with 600/100 mg bid than in PIs arm at week 24,63,64 48, 6 96, 65 and 144. 66
POWER 3 investigated efficacy and safety of DRV/r 600/100 mg bid in a larger population to support regulatory approval of darunavir. Inclusion and exclusion criteria of POWER 3 were similar to those of POWER 1 and 2 but in addition patients randomized in POWER 3 study were naïve to darunavir. HBV and HCV co-infected patients were included in the study as long as their condition was clinically stable and as long as they would not require any treatment for hepatitis infection during the trial period. After genotype resistance testing each patient received DRV/r 600/100 bid in addition to OBR with or without enfurvitide. The primary efficacy endpoint was the proportion of patients with at least -1 log10 reduction in viral load (using ITT analysis): and this was achieved by 65% of patients after 24 weeks and by 39% of patients at the final 144 week analysis.
Considering the secondary end point, the percentage of patients achieving a HIV/RNA < 50 copies/ml was respectively 40% and 32% at week 24 and 144 respectively. CD4 cell counts increased by a mean of 80 cells/mm3 at week 24 s and 118 cell/mm3 at week 144. In conclusion POWER 3 study confirmed that DRV/r 600/100 mg bid with an OBR was effective for treatment-experienced patients with multiple PI-resistance mutations.
TITAN study (TMC114/r In Treatment-experienced pAtients Naïve to lopinavir) is on-going, international, randomised, controlled, open-label 96 week phase III trial comparing efficacy and safety of DRV/r with lopinavir-ritonavir (LPV/r) in 595 treatment-experienced lopinavir-naïve patients. The inclusion criteria of the study were: HIV infected-patients at least 18 years old with viral load >1000 copies/ml. Patients should have been received a previous treatment with HAART for ≥12 weeks. Resistance test had to be done in all patients and used to assist the investigator in choosing an OBR. Exclusion criteria were AIDS–-defining illness (except stable Kaposi's sarcoma and wasting syndrome), clinically significant disease and clinical or laboratory evidence of decreased hepatic function. Previous or current use of lopinavir, darunavir, tipranavir, enfuvirtide or an investigational antiretroviral drugs was also exclusion criteria. Patients with chronic HBV and HCV were allowed to enter in the trial as long as their condition was clinically stable. At randomization patients received DRV/r 600/100 mg bid or LPV/r 400/100 mg bid associated to an OBR consisting at least two antiretroviral drugs (NRTIs with or without NNRTIs). Patients in the LPV/r arm were allowed and encouraged to switch to the 200/50 mg new tablet formulation. In patients receiving an NNRTIs in the OBR, the dosage of LPV/r was increased to 533/133 mg bid for the capsule and 600/150 mg bid for tablet formulation. 68 The primary efficacy outcome was to demonstrate non-inferiority of virological response (viral load <400 copies/ml) at week 48 of DRV/r versus the LPV/r arm (lower limit of 95% confidence interval [CI] for the difference in virological response [DRV/r-LPV/r] between the arms should not be greater than -12% in the per-protocol analysis). If non-inferiority was established, an a priori secondary end point was to demonstrate the statistical superiority of one arm over another (ITT population). Secondary end points were the proportions of patients with a viral load lower than 50 copies/ml and a 1.0 log10 or more reduction from baseline in viral load (TLOVR), and median CD4 cell count change over time. Safety was also assessed (ITT analysis). Rate of virological failure in LPV/r group was much higher than in DRV/r arm (11% vs. 1%) while the incidence of discontinuation due to adverse events (AEs) was similar between two groups. At week 48 more patients in the DRV/r than in the LPV/r had confirmed viral load of less than 400 copies/ml (77% vs. 68%, estimated least square mean difference 9%, 95% CI: 2–16) Since the lower limit of the 95% CI for the difference in response between treatment arm was nor lower than -12%, this result established non inferiority of DRV/r. At week 48 patients receiving DRV/r also had significantly higher responses than those receiving LPV/r for virological secondary end points and a similar increases in CD4 cell count was observed between treatment groups. TITAN study analysed the development of resistance in terms of virological failure. Among patients with virological failure, loss of susceptibility to PIs or NRTIs after treatement occurred in more patients in LPV/r arm than in DRV/r arm. Finally a recent analysis at week 96 showed the same results. 69
Therefore TITAN data confirmed that DRV/r was not only non-inferior to LPV/r but superior than LPV/r at 48 and 96 weeks.
The latest evidence has demonstrated the noninferior efficacy of a monotherapy regimen with DRV/r 800/100 mg once daily versus a triple combination therapy containing two nucleosides and DRV/r after 48 weeks of treatment. Nevertheless, it is still premature to consider switching to DRV/r monotherapy at this time. 70
Treatment-naïve adult patients
ARTEMIS is an international, randomized, open-label, 192 week Phase III trial conducted on 689 treatment-naïve adult patients. The enrolled participants were at least 18 years old, naïve to antiretroviral drugs and had a viral load greater than 5,000 copies. Patients were randomly assigned to receive 800/100 mg once-daily DRV/r or 800/200 mg LPV/r given once or bid, plus OBR with fixed-dose tenofovir/emtricitabine (Truvada coformulation). 31 Exclusion criteria were AIDS–-defining illness, clinically significant disease and clinical or laboratory evidence of decreased hepatic function, acute viral hepatitis, creatinine clearance less than 70 ml/min, pregnancy and breastfeeding women. Patients with chronic HBV or HCV entered in the trial as long as their condition was clinically stable. Patients were stratified according to baseline viral load (mean 4.85 log10 copies/mL) and CD4 cell count (median 225 cells/mm3). The overall discontinuation rate was low in both treatment group. In DRV/r group discontinuation due to AEs or due to virological failure occurred in 3% and <1/% of patients respectively. In LPV/r group discontinuation due to an AEs and due to virological failure occurred in 7% and 2% of patients respectively. At 48 week, 84% of DRV/r patients achieved a viral load l < 50 copies/ml versus 78% of patients treated with LPV/r. Thus DRV/r was non inferior to LPV/r in treatment- naïve patients. In patients with baseline viral load ≥100.000 copies/ml boosted darunavir was superior to lopinavir at week 48 and at week 96, 71 with a higher number of patients achieving plasma viral load <50 copies/ml. 31
In conclusion the trial showed that once-daily DRV/r is safe and effective in HIV treatment-naïve patients.
Treatment-experienced children and adolescents
On December 2008 the FDA announced the approval of the new 75 mg darunavir tablet for use by children with HIV aged from 6 years to 17 years old. 15 This indication is based on DELPHI (Darunavir EvaLuation in Pediatric HIV-1-Infected treatment-experienced patients) trial, an open-label Phase II study looking at the safety and efficacy of DRV/r in treatment-experienced pediatric patients aged 6 to 17 years. 72 The dose of the drug is based on the child's weight: the minimum weight required is 20 kg. Eighty patients (71% male), most vertically infected, were included in the study. The mean baseline viral load was 4.64 log10 copies/ml, the median CD4 count and percentage was 330 cells/mm3 and 17% respectively. All patients had been on HAART for at least 12 weeks. The patients had a median of 3 primary PI resistance mutations, 11 PI resistance-associated mutations, 2 NNRTI resistance mutations and 4 NRTI resistance mutation. All patients received an OBR in association with DRV/r. Pharmacokinetics, safety and efficacy were assessed throughout the follow up period of 48 weeks. At week 48, 73 65% of patients achieved at least a 1.0 log10 reduction of HIV RNA from baseline (TLOVR analysis); 59% achieved viral load <400 copies/ml and 48% an undetectable viral load and the mean CD4 cell increase was 147 cells/mm3. Fifty-nine % of patients with no baseline darunavir resistance mutations achieved an HIV RNA < 50 copies/ml compared to 47% of patients with 1–2 such mutations and 0% with 3 ore more mutations. According to this data DRV/r showed good virologic response rates in the presence of <3 DRV resistance-associated mutations. Thus, DRV/r seems to be a valuable therapeutic options for treatment-experienced pediatric patients.
The use off-label of new antiviral agent will be a compelling issue in the vertically HIV-infected infants born in the era of HAART. In our experience, 74 we treated a 21 months patient with vertically-acquired multidrug-resistance HIV infection and severe clinical and immunological stage of disease (CDC class C3) with DRV/r and etravirine. During the 15 months of follow up we showed that the administration of the two investigation drugs was safe and effective and drive a sustained quantitative and qualitative immune recovery. Thus we believe that the use of experimental agents with a careful follow up should be considered in infant with limited treatment options.
Ongoing trials
GRACE is multicentre, ongoing, open-label, phase IIIb study comparing the efficacy, safety and tolerability of DRV/r according to gender and race. 75 Two hundred eighty-seven women and 142 men with HIV infection, plasma HIV-RNA ≥ 1000 copies/ml were enrolled. Previous intolerance or failure with PI and/or NNRTI based HAART regimen of at least 12 weeks were an inclusion criteria. The primary outcome measure was to evaluate all differences, between women and men, in efficacy of DRV/r 600/100 mg bid plus an OBR over 48-week period. Secondary outcome measures were: change in viral load and CD4 from baseline; change in lipid profile and metabolic parameters, quality of life; changes of body shape and associated distress over 48 week study period. At week 24 the virological efficacy was similar in women and men (65.5% vs. 64.8%). No unexpected AEs were noted in interim analysis and the rate of AEs was not influenced by gender.
DESeRT study (Study in Healthy Males to Measure Darunavir and Etravirine in Blood, Seminal Fluid, and Rectal Tissue) is being conducted to look at how the body handles darunavir and etravirine in adult patients. 76 It will measure the amount of darunavir and etravirine in blood, semen, and in the rectum of men to search how much of the drug reaches the reproductive and intestinal tracts. The presence of the drug in these areas may be beneficial in preventing the sexual transmission on HIV virus.
Safety and Tolerability
The safety assessment of Darunavir is based on safety results obtained in adult in treatment experienced-patients included in POWER and TITAN studies and in treatment-naïve patients from included in ARTEMIS study. The complete list of adverse events is shown in Table 1.
Adverse events of Darunavir.
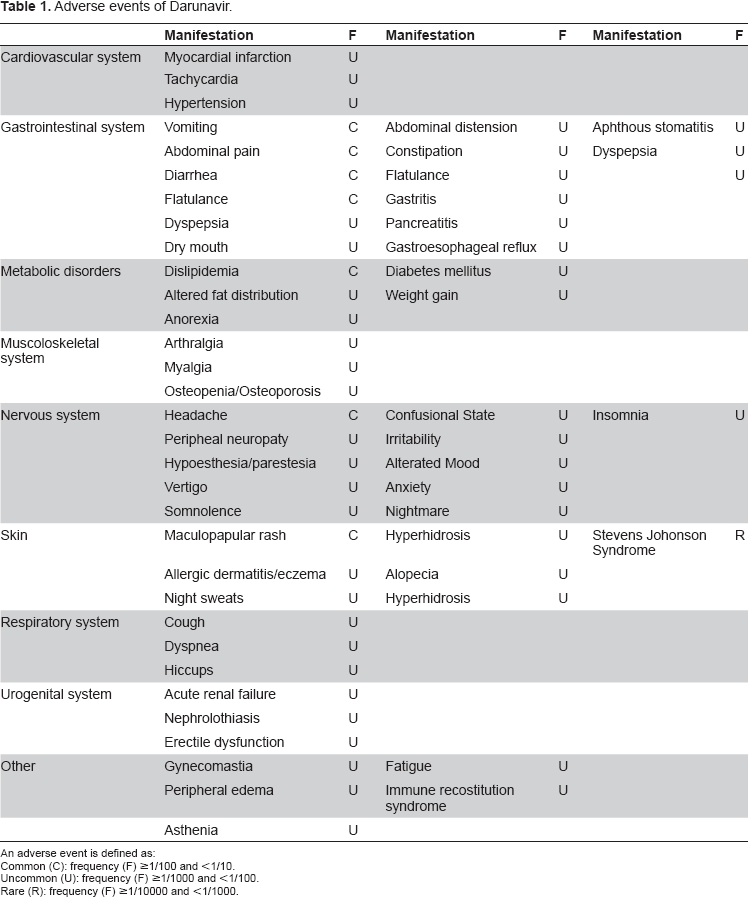
An adverse event is defined as:
Common (C): frequency (F) ≥1/100 and <1/10.
Uncommon (U): frequency (F) ≥1/1000 and <1/100.
Rare (R): frequency (F) ≥1/10000 and <1/1000.
Safety of Darunavir in treatment-experienced adult patients
Darunavir/r was well tolerated in POWER trials AEs were mild to moderate in severity (grade 1 or 2). There was a limited number of grade 3 and 4 adverse events. Excluding enfuvirtide-associated injection-site reactions, the most common adverse events at 48 weeks were diarrhoea (20% vs. 28%), nausea (18% vs. 13%), headache (15% vs. 20%) pharyngitis (14 vs. 11%), fatigue (12 vs. 17%), upper respiratory infection (12% vs. 7%) and herpes simplex (12 vs. 2%). 6 In darunavir group lower or similar incidence of this AEs was observed compared to control PI group. However when the difference in treatment exposure between the two treatment arm was taken into account, the analysis per 100 patients-years of exposure showed lower clinical adverse events rates with DRV/r. 6 The incidence of severe adverse events was low in both treatment group and did not lead to treatment discontinuation. Considering the changes in lipid values at 48 weeks, grade 3 and 4 laboratory abnormalities were observed in both treatment arms. Increased triglycerides (>8.4 mmol/1) was observed in 15% of darunavir treated patients vs. 7% of PI treated patients. Increased total cholesterol (>7.7 mmol/L) occurred in 7% of patients treated with DRV/r vs. 2% in patients in PI group. In DRV/r arm 6% of patients showed an increased level (>2 x upper limit of normal) of pancreatic amylase versus 5% of patients in PI group. The increasing in pancreatic lipase (>2 x upper limit of normal) was higher in PI group than in DRV/r group (5% vs. 1%). Grade 3 and 4 increases in alanine aminotransferase and aspartate aminotransferase (>5 x upper limit of normal) were observed in 2% and 4%, respectively, of patients in DRV/r group and 2% and 4%, respectively, of patients in control PI group. No cases of clinical pancreatitis and hepatotoxicity were reported. The overall incidence of laboratory abnormalities was similar between DRV/r and PI arms. No specific laboratory abnormalities were found to be associated with DRV/r treatment. At 144 week, 66 the combined analysis of POWER 1 and 2 showed that the most treatment-emergent adverse events from week 24 onwards in patients taking DRV/r 600/100 bid were diarrhea (16%), pharyngitis (14%), sinusitis (13%) and bronchitis (13%). Most adverse events were grade 1 or 2. The analysis of tolerability data from POWER 3, showed no new safety concerns.7,67 At week 144 the most common AEs were diarrhoea, vomiting and nausea generally grade 1 or 2 in severity; only 4% of patients reported grade 2–4 diarrhoea. Small changes from baseline, mainly grade 1 and 2 in severity, were observed in lipid profile (total cholesterol, triglycerides, HDL and LDL) and glucose. Treatment discontinuation due to AEs was relatively low (10%). 67 At 24 weeks POWER 1 and POWER 3 showed that in HIV patients with HB V or HCV, clinically stable, generally boosted darunavir treatment was well tolerated. 77 In this population, the administration of darunavir was associate with asymptomatic liver transaminase elevation in 13% of co-infected subjects vs. 8% in patients without coinfection.
In TITAN study DRV/r 600/100 mg bid was generally well tolerated at 48 weeks of treatment with similar types and incidences of adverse events to those observed in the POWER studies. 68 At week 48 the incidence of AEs was lower or similar with DRV/r than LPV/r The most frequent observed adverse events in DRV/r treated patients (grade 1 or 2 in severity) were diarrhea (32%), nausea (18%) and pharyngitis (12%), which occurred in 42%, 21% and 11.% of LPV/r patients, respectively. Rash-related events (regardless of severity and cause) were observed in 16% of subjects treated with DRV/r and in 7% in the LPV/r group. Grade 3–4 rash occurred infrequently in DRV/r group. The increased of total cholesterol was similar for patients in both treatment arm; however the mean percentage change in triglycerides from baseline was greater in patients in LPV/r group than in DRV/r arm. Elevation of liver transaminase was similar in the two treatment group. (Table 2) Discontinuation rates due to AEs were relatively low (7%) in patients receiving DRV/r. At the final 96-week analysis of the Phase III TITAN trial, darunavir showed a safety and tolerability profile similar to the data collected in the previous studies. 70 The incidence rates of treatment-related grade 2–4 diarrhoea were lower in subjects receiving DRV/r than LPV/r ones (8% vs. 15%) whereas rash was more frequently in darunavir treatment group (3% vs. 1%). Changes in triglycerides and total cholesterol were lower in DRV/r arm.
Adverse events and laboratory abnormalities of Darunavir in comparison to Lopinavir in TITAN and ARTEMIS study.
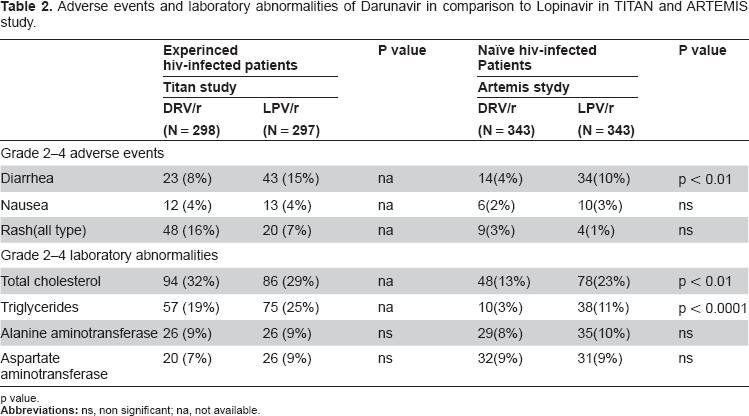
p value.
Safety of Darunavir in treatment-naïve adult patients
In treatment naïve-patients once-daily DRV/r was generally safe and well tolerated and the tolerability profile was similar to that reported in treatment-experienced patients. 31 Drug-related adverse events and laboratory abnormalities occurred in subjects treated with darunavir and were mild to moderate in severity. At 48 weeks the most common treatment-observed AEs were diarrhea, nausea, headache, upper respiratory tract infection, pharyngitis, abdominal pain, vomiting and cough (regardless of severity and causality). At week 48, patients on DRV/r were less likely to have moderate to severe (grade 2–4) treatment-related diarrhea than patients on LPV/r (4% vs. 10%; P < 0.001). The overall incidence of laboratory abnormalities observed at week 48 was similar in the two treatment group. Analysing the lipid profile, the mean increase in triglycerides and total cholesterol was higher in subjects treated with LPV/r than in DRV/r group; grade 2–4 elevation in triglycerides occurred more frequently in LPV/ arm (see Table 2). The incidence of grade 2–4 rash was similar in both groups. One case of Stevens-Johnson syndrome occurred in DRV/r group, a relationship with darunavir could not be excluded. Serious adverse events occurred in 7% of patients in DRv/r arm and in 12% in LPV/r arm. In this trial, rates of discontinuation of therapy due to adverse events occurred infrequently: in particular rates were lower in subjects receiving darunavir than in the LPV/r group at 48 weeks (3% vs. 7%) and confirmed at 96 weeks (4% vs. 9%). At 96 weeks, DRV/r (once-daily) continued to be associated with better safety assessment with lower rates of diarrhea, and increases in triglycerides and total cholesterol than boosted lopinavir.
ARTEMIS study shows that once-daily darunavir/ ritonavir offers a new, effective, well-tolerated once-daily first-line treatment option for treatment-naive patients.
Safety of Darunavir in treatment-experienced children and adolescents
Safety of Darunavir in pediatric treatment-experienced population has been evaluated on DELPHI trial at week 24 and week 48.73,74 DRV/r had a favourable safety and tolerability profile at week 24. This data were confirmed throughout the follow up period. At week 48 the most common treatment-emergent AEs were fever, cough, upper respiratory infections and diarrhea occurring at least in 15% of patients. Most AEs were mild to moderate (grade 1–2); only 26% of patients had a grade 3–4 AEs. Fourteen % of patients experienced serious AEs, but no deaths were reported, 1 patient discontinued therapy due to grade 3 anxiety considered unrelated to darunavir. Grade 2 and 4 laboratory abnormalities were observed in >1% patients as reported in Table 3. At week 48 there was a small but statistically mean increase in total cholesterol, LDL and HDL, however no statistically changes in total cholesterol/HDL ratio were seen in patients treated with DRV/r. The mean triglyceride level decreased significantly. No significant changes were seen in glucose metabolism. 78
Laboratory Abnormalities at week 48 in DELPHI trial.

FDA safety labelling revisions
In October 2008 the FDA approved safety labelling revisions for darunavir tablets warn of the adverse events, drug interactions and pregnancy risk. Based on the results of clinical trials and postmarketing surveillance. 15 DRV/r induces hepatitis therefore liver function should be monitored before and during treatment (especially during the first months) in patients with chronic hepatitis, cirrhosis or conditions characterised by elevated pre-treatment levels of transaminase. Darunavir should be discontinued if severe rash develops. Cases of Stevens-Jonhnson Syndrome have been reported. New-onset diabetes mellitus or hyperglycemia, redistribution or accumulation of body fat, immune reconstitution syndrome or increased bleeding events in patients with haemophilia occur during darunavir treatment. Darunavir should be used with caution in patients allergic to sulfonamides.
Special populations
The safety profile of Darunavir was evaluated in the special populations reported below:
Hepatic Impairment
Patients with Renal Impairment
Pregnant and Breast-feeding women
Elderly Patients
According to the pharmacokinetic data, the concentration-dependent protein binding (AAG) of darunavir should be considered in patients with impaired hepatic function. The free fraction of darunavir may be increased in patients with mild and moderate hepatic dysfunction (Child-Pugh class A or B) due to lower AAG: this fact is considered an issue for the safety. Therefore darunavir should be used with caution in impaired hepatic function but dosage adjustment is not necessary. 16
The use of darunavir is contraindicated in patients with severe hepatic impairment (Child-Pugh class C).
Pharmacokinetics of darunavir is not significantly affected in patients with moderate renal function impairment (Creatinine Clearance of 30 to 60 mL/min) because of predominant liver metabolisation. However data are not available for patients with HIV and severe renal function impairment or end-stage renal disease. Anyhow, dosage adjustment is not required in patients with renal impairment.
The use of darunvir during pregnancy is admitted only if the potential benefit justifies the potential risk according to FDA classification (category C). There are no adequate and well-controlled studies conducted in pregnant women. An Antiretroviral Pregnancy Registry has been established to monitor the maternal-fetal outcomes of pregnant women exposed to darunavir. 16 It is not known whether darunavir is excreted in human milk or not; however data show that it is excreted in the milk of lactating rats. Because of the potential for HIV transmission and for serious adverse effects from darunavir to the breastfed infant, women should not breastfeed while taking darunavir. 16
Limited information is available on the use of darunavir in patients aged >65 years. However, elderly patients are more likely to have age-related liver problems, concomitant disease or other theraps which may require caution in administering darunavir. Race does not affect darunavir pharmacokinetics.
Quality of Life
In recent years an increasing attention has been devoted to the quality of life of HIV-infected patients treated with antiviral agents. Dubois et al analyzed the potential impact of DRV/r 600/100 mg bid on patients health-related quality of life (HRQoL). 79 This study was based on the analysis of the Functional Assessment of HIV Infection (FAHI) scores from the pooled POWER 1 and POWER 2 data. All patients included in POWER 1 and POWER 2 trials received a validate HRQoL questionnaire (FAHI): an established disease-specific instrument for the assessment of HRQoL in HIV/AIDS patients composed by 5 subscales. For each of the 637 patients a total FAHI score was calculated as the sum of the 5 subscale score. Higher score reflected a better quality of life. HRQoL questionnaire was administered at baseline and at week 4,12,24 and 48. At 48 weeks, patients treated with DRV/r 800/100 once daily 400/100 bid and 600/100 bid had significantly higher total FAHI scores than control PIs group; in particular a sustained, significant and clinically meaningful improvement in HRQoL was observed in DRV/r 600/100 bid compared to PIs group. These results are similar to the clinical findings established in POWER 1 and POWER 2. 79
Place in Therapy
Darunavir is the most recent PI retaining virological activity in the presence of multiple protease mutations. Thus, it appears to be useful component of optimized combination antiretroviral therapy for HIV-infected patients previously treated with antiretrovirals.
Darunavir was generally well tolerated in treatment experience and naïve HIV-1 infected patients.
As AEs are one of the major cause of nonhaderence leading to subsequent viral resistance, darunavir great safety profile has met quality requirements in order to improve therapeutic adhesion and quality of life and to maintain good virology efficacy. Recently, once-daily darunavir boosted–-ritonavir has been added as a component of the listed drugs in the preferred PI-based regimens in treatment-naïve adult patients in the US Department of Health and Human Services (DHHS) guidelines. 80
Moreover, darunavir has been approved in children and adolescents ranging 6 to 17 years of age. The evidence shows that darunavir is a potent and well tolerated option for treatment of highly antiretroviral-experienced children and adolescents and they closely match the efficacy reported in treatment-experienced adults. However, data on the therapeutic use for naïve children and adolescents as well as for children younger then six years are still missing. 71 Nevertheless, newly updated sections in DHHS for antiretroviral therapy in children and adolescents include darunavir as a valuable choice for salvage therapy in this population. 81
Vertical transmission of multi-drug resistant HIV although rare, may occur.74,82,83 Darunavir efficacy has been described in one case of perinatal transmission of multidrug-resistant HIV type-1), 74 suggesting that the experimental use of this agent might become a compelling issue in vertically HIV-infected children born in the era of HAART.
Conclusion
Darunavir is considered and validated as an important partner of an optimized nucleoside in reverse transcriptase back bone in the treatment of heavily pre-treated adult patients. Owing to its good safety and efficacy profile it has acquired greatest relevance as a treatment option among naïve HIV infected adults and treatment experienced HIV-infected children and adolescents. Further studies are needed to provide information for its use in children younger then 6 years. New evidence has proposed its possible use as a monotherapy regimen, the datum needs to be further confirmed.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors report no conflicts of interest.