
Abstract
This article reviews the role of vernakalant in the management of atrial fibrillation (AF). Published data in English language were identified from MEDLINE and Current Content database (both 1966 to January 30, 2009). Vernakalant is an antiarrhythmic agent with sodium and ultra-rapid potassium channel blockage property and atrial selective effect. In clinical studies evaluating intravenous vernakalant in cardioversion of patients with recent onset AF, vernakalant improve the chance of acute restoration to normal sinus rhythm (NSR). In post-operative AF, the chance of conversion to NSR was also improved. Phase II studies demonstrated that oral vernakalant 300 mg or 600 mg twice daily successfully maintained normal sinus rhythm compared to placebo. Common side effects include dysgeusia, sneezing, and paresthesia. Future studies are needed to explore the efficacy and safety of using vernakalant with patient populations who are prone to AF, as well as its comparative efficacy and safety to other antiarrhythmic agents.
Introduction
Atrial fibrillation (AF) is a common type of atrial arrhythmia and chronic AF doubles the risk of total and cardiovascular mortality. Risk of AF increases with age with an incidence up to 26.0% for men and 23.0% for women by the age of 40. 1 In patients undergoing cardiothoracic surgery, the risk of AF post-operation can be as high as 20%-50%. 1 Management of AF involves achieving 3 major goals, ventricular rate control (rate control), preventing thromboembolic events and restoration of normal sinus rhythm (NSR) (rhythm control). 2 Rate control involves the use of atrioventricular nodal blocking agents such as beta-blockers, calcium channel blockers (diltiazem and verapamil) or digoxin. Patients who receive rate control management strategy will require long-term anticoagulation with warfarin therapy to prevent thromboembolic events. Except if they only have one of the following risk factor: congestive heart failure, hypertension, age >75 years, diabetes and previous thromboembolic events, then aspirin may be used. Rhythm control involves the use of Class Ia, Ic and III antiarrhythmic agents such as procainamide, flecainide and amiodarone, for restoring and maintaining NSR. 2 Theoretically, once NSR is restored, anticoagulation therapy can be discontinued after a month. 2 However, in certain high risk patients, recent study evaluating rate versus rhythm control strategy indicated that long-term anticoagulation therapy may also be needed. 3 In recent years, numerous clinical trials have compared the rate control strategy versus rhythm control strategy in long term cardiovascular outcomes in patients with AF. From a therapy point of view, each strategy has its pros and cons. 4 Overall, rate and rhythm control strategies are believed to produce non-significant differences in long-term cardiovascular outcomes.
The rhythm control agents (i.e. Class Ia, Ic and III antiarrhythmics) that are currently available on market have variable efficacy and safety limitations. 5 By targeting myocardial ion channels responsible for impulse propagation in cardiac tissue, or prolonging effective refractory period as they attempt to restore sinus rhythm by breaking up the multiple wavelets of electrical activity that sustain fibrillation, they lack selectivity to the atrial tissue.6,7 As they affect the ventricular tissue concurrently, these agent can induce proarrhythmic events (Torsades de Pointes [TdP] or monomorphic ventricular arrhythmia), especially in high risk patients such as those with other underlying cardiovascular diseases.8,9 Rhythm control agents have been reported to double the risk of TdP in all patients. In certain high risk patients such as those with heart failure, the use of rhythm control agent may increase the risk of TdP up to 6 times higher. 9
New development in antiarrhythmic agents these days focus on minimizing the risk of TdP. Vernakalant (Kynapid™, RSD 1235, by Cardiome Pharma Corporation and Astellas Pharma Inc. in the U.S.), is a new antiarrhythmic agent that has been studied for rhythm control in AF. Unlike other agents on market, vernakalant is a mixed blocker of cardiac K+ and Na+ channels and it demonstrates atria-selective action and only minimally affects ventricular repolarization, thus potentially minimizing the risk of TdP. 10 The intravenous (IV) form of vernakalant used for acute cardioversion was submitted to the U.S. Food and Drug Administration (FDA) and was reviewed on December 11, 2007, whereas the oral form of vernakalant is under Phase II investigation for maintenance of NSR. The Cardiovascular and Renal Drugs Advisory Committee had voted in favor of the FDA approving Vernakalant. 11 Final decision has not yet been made to-date. On August 8, 2008, Astellas Pharma received a letter from the FDA stating that they would require additional information associated with the risk of previously identified adverse events during the clinical trials in order to assure an acceptable risk benefit ratio compared to electrical cardioversion. FDA also requested a safety update from ongoing or completed studies. Astellas is in the process of addressing these concerns. This article reviews the pharmacology and clinical evidence available to-date of the use of vernakalant in the management of AF, as well as its potential place in future management of AF.
Mechanism of Action, Metabolism and Pharmacokinetic Profile
Atrial fibrillation is caused by reentrant wavelets propagating in atrial-tissue. 12 Inward sodium current (INa) is responsible for the rapid upstroke of the action potential. Class Ia (procainamide, quinidine and disopyramide), and Ic (flecainide, propafenone) antiarrhythmic agents block INa, which interrupt arrhythmic circuits by reducing impulse conduction velocity and tissue excitability. 13 Their potency is rate-dependent, which means there will be greater degree of inhibition of the INa during AF, when the atrial rate is faster and frequency of atrial depolarization is high. 14 Some class I antiarrhythmic agent such as propafenone also exhibit voltage dependent INa blockage. This means the drug exerts more potent blockade of INa when the tissue is depolarized, as in AF. Therefore, antiarrhythmic agents that have voltage and frequency dependent INa blockade property may have minimal effect on normal tissue and produce selective antiarrhythmic action during AF. 14 Class III agents (sotalol, amiodarone, dofetilide) inhibit both rapidly (IKr) and ultra-rapidly activating (IKur) delayed rectifier potassium current. This increases action potential duration and enhance refractoriness, ultimately extinguishing atrial fibrillation. 15 Table 1 summarize the pharmacological profile of antiarrhythmic agents currently available on the US market.
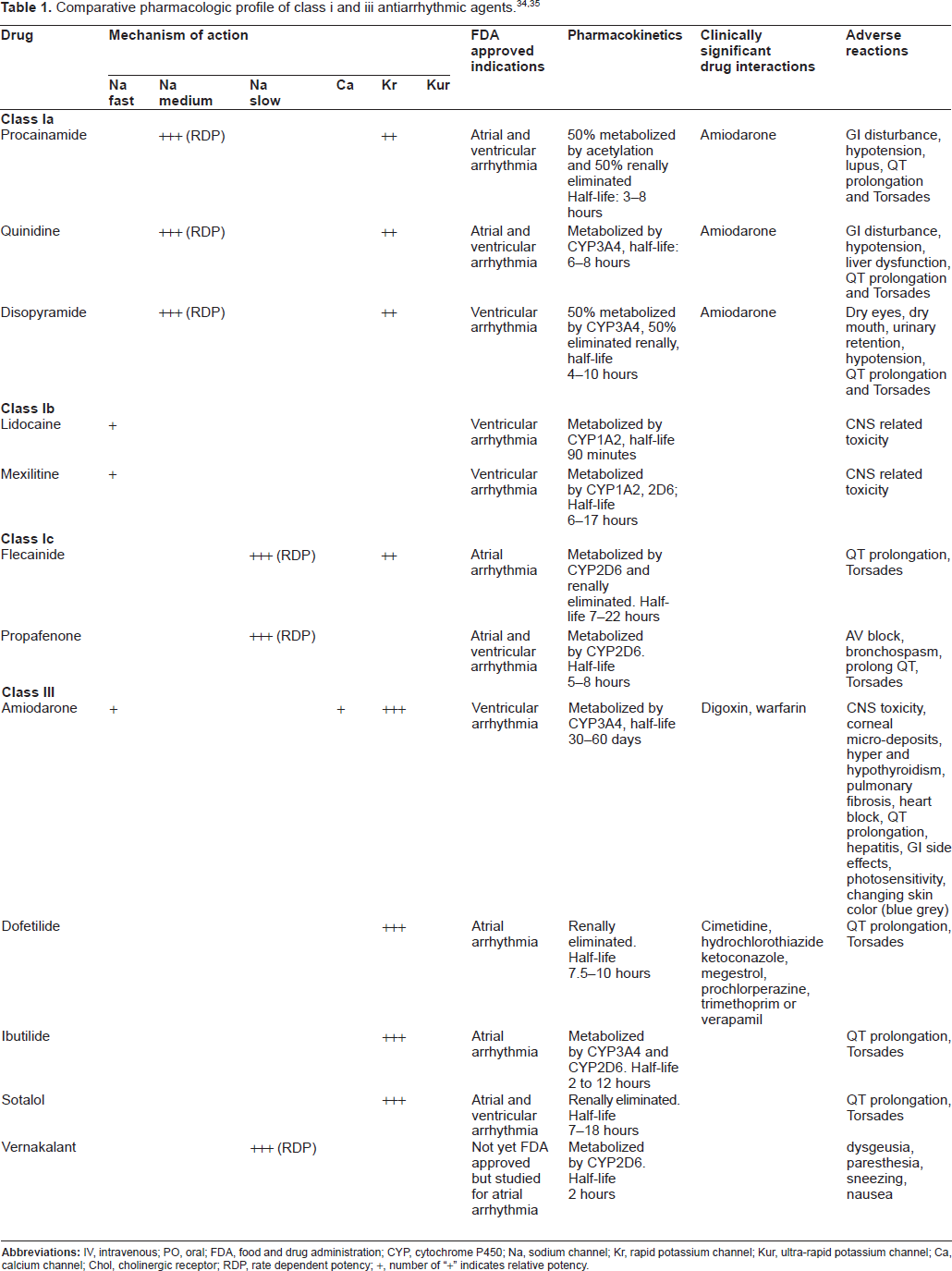
However, since INa and IKr are present in both atrial and ventricular tissue, antiarrhythmic agents that inhibit either of these channels will prolong repolarization of both the atrium and the ventricles. Prolongation of ventricular repolarization increases the risk of TdP. IKur on the other hand, is thought to be atrial specific in human heart. 16 The protein that contributes to the formation of IKur, Kv1.5 protein is only found in human atrial but not ventricular myocytes. 17 There are currently no antiarrhythmic agents available that specifically inhibit only IKur. Researches in new antiarrhythmic agents focused on increasing the drug's atrial selectivity and minimizing side effects such as TdP.
Mechanism of Action of Vernakalant
Vernakalant (3-pyrrolidinol, 1-[(1R,2R)-2-[2-(3,4-dimethoxyphenyl)ethoxy] cyclohexyl]-, hydrochloride (3R)-) is a new antiarrhythmic agent currently being studies and awaiting FDA approval. It is a rate and voltage dependent INa inhibitor, as well as an atria selective IKur and Ito inhibitors. 18 In non-polarized tissue, vernakalant exerts no INa and IKur inhibitory effect. In normally polarized tissue, vernakalant blocks Na+ and ultra-rapid rectifier K+ currents. 18 During AF, this mechanism of action allows the drug to bind of Na+ channel in the rapidly activating and depolarized atrial tissue rather than to the ventricles which are beating in slower rate relative to the atrium. It has been demonstrated that vernakalant has minimal prolongation in QT and QRS interval at peak plasma concentrations used clinical study dosages (2-3 mg/kg). This indicates that ventricular repolarization time and ventricular conduction velocity are only minimally affected by vernakalant. 19 In addition, it has been demonstrated that in rabbit model, vernakalant prevented the action potential prolongation caused by dofetilide and terminated TdP induced by clofilium, a class III antiarrhythmic agent. 18 Furthermore, it is now known that sodium current in the heart is a mixture of currents with different kinetics. 20 The combination blockade of the I(Na(P)), a very slowly inactivating component of the sodium current and the K channel may have also provided protection from causing TdP in vernakalant.
Pharmacokinetics
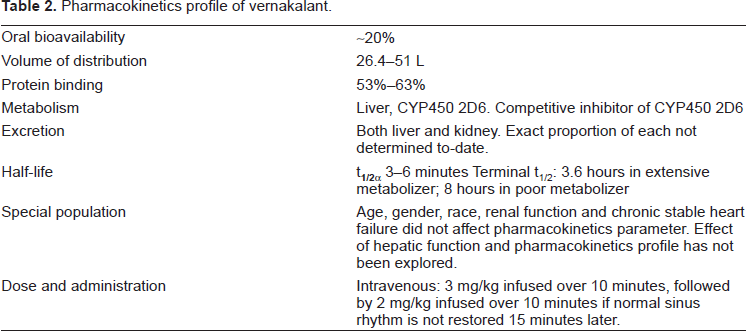
The pharmacokinetics profile has been explored in normal volunteers as well as patients with AF or atrial flutter. Mao and colleagues studied the pharmacokinetics of vernakalant in 128 patients with AF or atrial flutter.21,22 The majority of patients are male (70%) and Caucasian (92%). Twenty percent of patients have history of chronic stable heart failure. Thirty-seven percent of study patients had mild renal insufficiency (creatinine clearance of 50-79 ml/min) and 12% had moderate renal insufficiency (creatinine clearance of 30-49 ml/min). Median serum albumin and serum bilirubin concentrations were 4.1 g/dL (range, 2.9-5.2 g/dL) and 0.6 mg/dL (range, 0.1-2.1 g/dL), respectively. Following two intravenous infusions of 3.0 mg/kg and 2.0 mg/kg, each over 10 minutes and separated by 15 minutes if NSR is not restored, vernakalant's pharmacokinetics was described by a two-compartment model with first order elimination, from the central compartment (with a short first distribution phase: alpha-phase half-life of 3-6 minutes). 21 The maximum plasma concentration, area under the plasma concentration curve and elimination half life are found to be 3.29 mcg/ml, 11.64 mcg.hr/ml, 3.1 hour in men and 4.57 mcg/ml, 11.64 mcg.hr/ml, 2.9 hour in women respectively. Age, race, renal insufficiency, hepatic impairment, chronic heart failure did not demonstrate significant effect on the pharmacokinetic profile of vernakalant. Vernakalant is cleared both by the liver and the kidney. However, hepatic and renal clearances have not been determined separately. CYP4502D6 is the predominant enzyme involved in the O-demethylation metabolism of vernakalant. 21 The inhibitory potential of vernakalant was very weak for cytochrome P450 1A2, 2C9, 2C19, 2E1, and 3A4. In vitro studies suggested that vernakalant is neither a reversible nor irreversible inhibitor of the above cytochromes, and is a moderate and competitive inhibitor of CYP2D6. 21 The mean steady state volume of distribution of vernakalant was found to be 85.84 L for extensive metabolizers and 112.50 L for poor metabolizers. In poor CYP2D6 metabolizers, vernakalant clearance was 64% of that in extensive metabolizers, but Cmax did not differ. The estimated terminal population half-life was 3.2 and 8 hrs for CYP2D6 extensive and poor metabolizers, respectively. The volume of the central compartment was estimated to be 51.0 L and 26.4 L in male and female, respectively. However, when adjusted to body weight, the gender difference was not significant. 21
In a phase I, single-blind, ascending-dose trial, 29 healthy volunteers were randomized to receive either increasing doses of IV infusion of vernakalant (0.1-5 mg/kg) or placebo over 10 minutes. Vernakalant demonstrated linear pharmacokinetics in this dose range. 21 The maximum plasma concentration of vernakalant after this single dose administration ranged from 0.08-4 μg/ml. Half-life of vernakalant in healthy volunteers is 2 hours. 19
Data from another phase I pharmacokinetics study of oral vernakalant in healthy volunteers observed that dose dependent increase in plasma concentration was seen and steady state concentration was achieved in 4 days. 19 The maximum dose given for 7 days was 900 mg twice daily (1,800 mg/day), yielding blood levels of vernakalant approaching peak blood levels seen in IV dosing (2 to 3 mg/kg) used in clinical trials. This indicated that the oral bioavailability of vernakalant is ~20%. The formulation provided sustained blood levels of drug over a 12 hour interval. 19 The plasma protein binding of vernakalant in human plasma is approximately 53% to 63% at therapeutic concentrations. 21
In a Phase II, multi-centered, randomized, double-blinded, step-dose, parallel group study, the pharmacokinetics of vernakalant was studied in 56 patients who have AF for a duration of 3 to 72 hours. 23 Patients were randomized to receive a 3 mg/kg vernakalant infusion administered over 10 minutes followed by a 2mg/kg infusion if restoration of NSR did not occur in 15 minutes, or a 1 mg/kg infusion, followed by a 0.5 mg/kg infusion in 15 minutes if NSR was not restored. The mean peak plasma concentrations were 5.8 μg/ml in the patient who received the 3 and 2 mg/kg infusion and 1.9 μg/ml in those who received both the 1 and 0.5 mg/kg infusion. The mean terminal elimination half-life in these patients was 3.1 hour (1.7-5.4 hour). 23
Table 2 summarizes the pharmacokinetics parameters and dosing information for vernakalant. Overall, vernakalant obeys 2 compartment pharmacokinetics with a short distribution phase with a half-life of 3-6 minutes and an elimination phase half-life of 3.6 to 8 hours depends on if patient is an extensive CYP450 2D6 metabolizer or a poor metabolizer. Whether the rate of CYP2D6 metabolism has any effect on clinical efficacy and tolerability of vernakalant has not been studied. The manufacturer of vernakalant has not made any recommendation of routinely checking patients’ metabolism status before drug administration. Currently, vernakalant is under consideration for approval for use in acute cardioversion and patients receiving the drug will only be receiving two doses of vernakalant. However, if oral vernakalant eventually also receive approval, the effect of different metabolism status will be more important to clinical outcomes as the patients will be on chronic therapy and that poor metabolizers may be more prone to accumulation of drugs.
Pharmacokinetics profile of vernakalant.
Pharmacodynamics
In a phase I, single-blind, ascending-dose trial of 29 healthy volunteers randomized to receive either increasing doses of IV infusion of vernakalant (0.1-5 mg/kg) or placebo over 10 minutes. No effect on blood pressure and heart rate was observed. However, there was an increase in PR (peak increase of 23 msec for the 4 mg/kg group at minute 9 of the infusion and 28 msec for the 5 mg/kg group at 3 minutes after the end of the infusion), QRS (peak increases of 8 msec at minute 8 of the infusion and 11 msec at end of infusion for both the 4 and 5 mg/kg groups) and QT intervals (peak increases of 48 msec at minute 7 of the infusion for the 4 mg/kg group and 63 ± 10 msec at the end of the infusion for the 5 mg/kg group).19,21
Dorian et al. performed a pharmacodynamic study evaluating the atrial electrophysiology of vernakalant in human. 24 Nineteen patients who had clinical indication for diagnostic electro-physiology study and/or radiofrequency ablation for arrhythmia were enrolled. Electrophysiology studies were performed before and after the administration of one of two IV regimens of vernakalant [1) 2 mg/kg infused over 10 minutes followed by 0.5 mg/kg/hr infusion for 35 minutes; 2) 4 mg/kg infused over 10 minutes followed by 1mg/kg/hour infusion for 35 minutes]. The lower dose significantly prolonged the atrial refractory period (AERP) at 600 of pacing cycle length (p < 0.05), but not at 400 or 300 msec of pacing cycle length. The higher dose significantly prolonged AERP from 203 to 228 msec at 600 msec of pacing cycle length, from 182 msec to 207 msec at 400 msec of pacing cycle length, and from 172 to 193 msec at 300 msec of pacing cycle length (p < 0.05). There was no significant prolongation of ventricular refractory period at any doses of vernakalant. QT intervals were unchanged from baseline.
A study designed with primary objective of examining the effect of vernakalant on QTc intervals has not been performed. In clinical trials, vernakalant was found to prolong the QTc interval by 20 and 23 msec after vernakalant doses of 3 mg/kg and 3 + 2 mg/kg, respectively. 21 After a single 3 mg/kg dose, the change in QTc intervals was predicted to return to baseline within 6 hours for CYP2D6 extensive metabolizers and within 12 hours for poor metabolizers, based on the half-life of vernakalant in these two population (extensive metabolizer 3.6 hours, poor metabolizer 8 hours). After the 3 + 2 mg/kg regimen, mean change in QTc interval was predicted to return to baseline after 12 and 24 hours for extensive and poor metabolizers, respectively. 21 The manufacturer of vernakalant and the clinical pharmacology reviewer from the FDA suggested that ECG monitoring should be continued for at least 6 hours after use of vernakalant until the QTc is within normal limits for CYP2D6 extensive metabolizers, and 12 hours for CYP2D6 poor metabolizers receiving 3 mg/kg. 21 Since it is not routine clinical practice to check the CYP2D6 metabolism status in every patients, it would be prudent to monitor all patients’ QT intervals for 12 hours after vernakalant discontinuation.
Pharmacokinetics in Special Population
According to data on file with the company Astella Pharma, differences in age, gender, race and renal function do not affect pharmacokinetics profile of vernakalant. 21 The effect of liver function on the pharmacokinetics profile of vernakalant has not been explored to-date. History of chronic stable heart failure also did not significantly affect the pharmacokinetics parameters of the drug. 21 It is important to consider that current pharmacokinetics parameters were determined based on patients receiving two doses for acute termination of AF. With more study and clinical data available for oral vernakalant for long-term use, the degree of significance of organ dysfunction and vernakalant pharmacokinetics may be altered. The pharmacokinetics of vernakalant has not been studied in pediatric population.
Drug Interactions
The drug interaction profile of vernakalant has not been formally explored by the manufacturer. In addition to the pharmacokinetic study by Moe and colleague indicating that in patient receiving beta-blockers, calcium channel blockers and cytochrome P450 2D6 inhibitors, the pharmacokinetic profile of vernakalant was not affected, 22 two of the Phase III clinical studies on the use of IV vernakalant have included a small number of patients who concurrently received vernakalant and digoxin (combined, n = 30) or who received concomitant class I antiarrhythmics (combined, n = 25), sotalol (combined, n = 28), or other class III antiarrhythmics (combined, n = 16).25,26 Coadministration of digoxin, class I antiarrhythmics, or class III antiarrhythmics was associated with NSR conversion rates of 20%, 20%, and 38%, respectively but not different from patients with no concomitant antiarrhythmic agents.
Clinical Studies
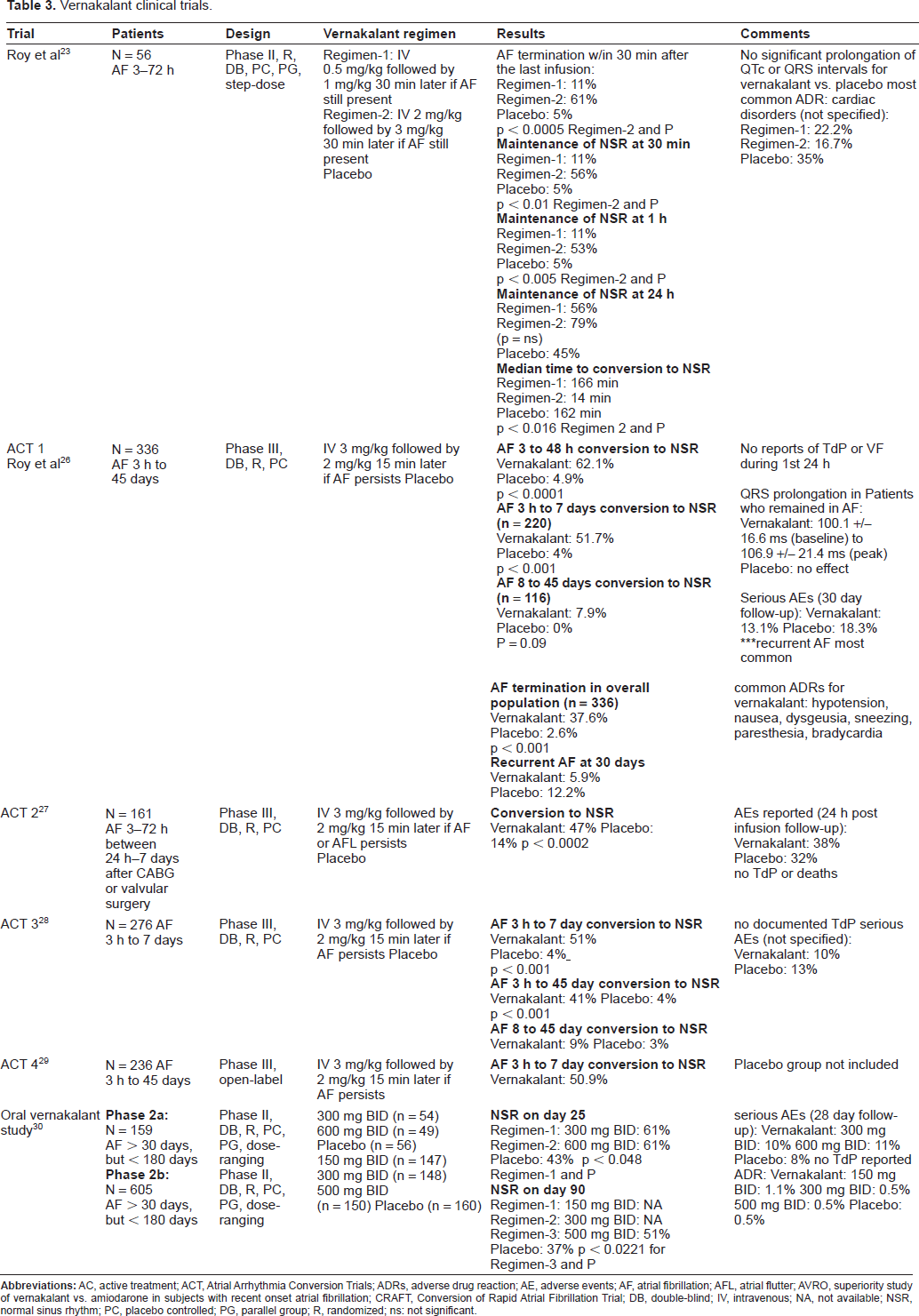
The summary of all vernakalant's clinical studies evaluating efficacy that were available at the time of writing are listed in Table 3. Intravenous vernakalant was assessed for the acute conversion of AF in one Phase 2 study and four phase 3 studies. Oral vernakalant was investigated for the cardioversion of AF and long-term maintenance of NSR in two Phase 2 trials. This section will discuss the clinical trials and its efficacy results. Please refer to the safety section for detail adverse events reported in each trial.
Vernakalant clinical trials.
Acute Conversion of AF to NSR
The efficacy and safety of IV vernakalant dosing was first explored by Roy et al in the management of recent onset AF. 23 This was a Phase II, multicenter, randomized, double-blinded, step-dose, placebo control, parallel group study. The trial included 56 patients with a recent onset of AF (new or recurrent AF lasting for 3 to 72 hours). They were randomized to receiving vernakalant 0.5 mg/kg followed by 1 mg/kg 30 minutes later if AF was still present, or vernakalant 2 mg/kg followed by 3 mg/kg 30 minutes later if AF was still present, or placebo. Patients were allowed to use beta-blocker, calcium channel blockers or digoxin for ventricular rate control at the discretion of their primary care physicians or cardiologists. Overall, approximately 68% of patients were on beta-blockers, 28% were on calcium channel blockers and 22% were on digoxin. The use of Class I and III antiarrhythmic agents preenrollment into the study was an exclusion criteria and the use of these agents was discouraged until 12 hours after vernakalant administration. The authors did not specify anticoagulation strategy used in this study population. The primary endpoint was termination of AF for any length of time during infusion or within 30 minutes after end of last infusion. Out of the three different treatment groups only the 2 + 3 mg/kg vernakalant group demonstrated a statistical significance difference in efficacy when compared to placebo in terminating AF (61% vs. 5%; p < 0.0005), maintenance of sinus rhythm at 30 minutes (56% vs. 5%; p < 0.01) and maintenance of sinus rhythm at 1 hour (53% vs. 5%; p < 0.005). The percentage of patients in NSR at 24 hours after the infusion was 79% in the 2 + 3 mg/kg vernakalant group, 56% in the 0.5 + 1 mg/kg group and 45% in the placebo group, indicating that patients continued to cardiovert after the infusion of drug ended. This may be due to a delayed respond to drug therapy or spontaneous conversion to NSR in patients. The median time to conversion to sinus rhythm for vernakalant 2 + 3 mg/kg was 14 minutes versus 162 minutes for the placebo group (p = 0.016). The investigator concluded that IV vernakalant in dosage of 2 + 3 mg/kg was efficacious in cardioverting recent-onset AF to NSR when compared to a lower dose of vernakalant (0.5 + 1 mg/kg). The incidence of adverse events was similar between treatment groups; however, cardiac events such as premature ventricular beats and non-sustained ventricular tachycardia were more common in placebo recipients. No drug-related pro-arrhythmias were reported. Although the number of subjects in this study was too small to determine the true tolerability of vernakalant, it provided information on the effective dose to be tested in Phase III study.
There are four Phase III studies evaluating the efficacy and safety of IV vernakalant on acute conversion of AF (The Atrial Arrhythmia Conversion Trials [ACT] 1 to 4). Preliminary results of ACT 1 and 3 were released in December 2004 and September 2005 respectively (full results of ACT 1 have been recently published). Results of ACT 2, a study evaluating patients with post-operative atrial arrhythmia, were released in June 2007. ACT 4, an open-label safety study evaluating recent onset AF, has recently finished patient enrollment.21,25,27,28
The recently published ACT 1 was a randomized, double-blinded, placebo controlled study designed to assess the effectiveness and safety of vernakalant in the conversion of AF. 26 Three hundreds and thirty-six patients with AF up to 45 days in duration were randomized to receive vernakalant 3 mg/kg infused over 10 minutes or placebo. A second dose of 2 mg/kg was given 15 minutes later if AF was not terminated. The sequence of the doses was reversed compared to the dose that demonstrated efficacy in Phase II study based on the assumption that more patients would convert if the higher dose was given first and that the Phase II study demonstrated safety of using 3 mg/kg. 21 Treatment with beta-blockers, calcium channel blockers or digoxin for ventricular rate control was permitted for up to 2 hours before the study drug administration. Treatment with other class I and III antiarrhythmic agents was allowed up to 24 hours before study drug infusion. No specification on the anticoagulation strategy use in this study population was made. Patients were stratified by duration of AF into 2 study groups: short-duration AF (3 hours to 7 days) and long-duration AF (8 to 45 days) and then randomized in a 2:1 ratio to receive vernakalant or placebo. Primary end point was restoration of NSR within 90 minutes. In the primary efficacy analysis, 51.7% of vernakalant patients in the short-duration AF group converted to sinus rhythm within 90 minutes compared to 4.0% of placebo patients (p < 0.001). Patients with acute AF (lasting 3 to 48 hours) demonstrated the highest conversion rate after vernakalant administration (62.1% vs. 4.9% with placebo; p < 0.001). The median time of AF termination was 11 minutes and the majority (76%) achieved it after receiving only the first infusion. Overall, in the short- and long-duration AF groups, statistical difference was reached with 37.6% of vernakalant patients experiencing AF conversion to sinus rhythm compared to just 2.6% of placebo patients (p < 0.001). Transient alteration in taste, sneezing, paresthesia and hypotension were the most commonly reported adverse reactions with vernakalant. One episode of TdP occurring 32 hours after vernakalant administration was reported in the 90-year-old woman. Recurrent AF requiring hospitalization was the most common serious adverse event during this study (placebo, 12.2%; vernakalant, 5.9%). The investigators concluded that vernakalant demonstrated rapid conversion of short-duration AF (3-hours to 7-days) to NSR with good tolerability. It is important to point out that a low number of patients with heart failure were included in this study (only 14% of patients with New York Heart Associaton NYHA class I–III) which limits extrapolation of the vernakalant's efficacy results to that particular patient population.
The ACT 3 study also evaluated the safety and efficacy of intravenous vernakalant in the conversion of AF to NSR in 276 patients with recent onset AF. 27 The study design of ACT 3 is similar to ACT 1. The recent-onset AF treatment group (3 hrs to 7 days) in this study had a significantly higher response rate in converting to NSR as compared to placebo (51% vs. 4%; p < 0.001; median conversion time was 8 minutes). Similar outcomes were also reached by the overall AF study population (3 hrs to 45 days) which had a 41% termination rate of AF vs. 4% of placebo patients (p < 0.001). In the longer-term AF population (8 to 45 days) the difference in termination rate of AF was not statistically significant.
A pooled analysis of patients from the ACT 1 and 3 studies demonstrated that NSR was achieved in significantly more patients given vernakalant injection than those given placebo (51.1% vs. 3.8%; p < 0.0001).21,25 It was noted that the use of beta blockers or calcium channel blockers as background rate control agents, did not affect AF-to-sinus rhythm conversion rate with vernakalant, but vernakalant was slightly more efficacious in conversion to sinus rhythm in patients taking sotalol as background rhythm control therapy and less efficacious in those taking digoxin or other class I or III antiarrhythmics as background therapy. Another pooled post hoc analysis showed similar efficacy of vernakalant in patients with ischemic heart disease, hypertension, and/or hepatic impairment as in those without; however patients with heart failure, renal impairment or those aged over 75 years achieved NSR less commonly. 26 The real clinical significance of the results from both sub-analyses is limited due to a small number of subjects being evaluated in the studies.
ACT 4 is an open-label phase 3 study designed to evaluate the safety of IV vernakalant and provide further evidence of efficacy in the conversion of AF to sinus rhythm. 21 Out of the 236 patients enrolled in this study, 167 had AF for more than 3 hours but less than 7 days (short-duration AF). The primary vernakalant efficacy endpoint (achievement of NSR in 90 minutes) was achieved in 50.9% of patients treated with vernakalant. The median time to conversion was 14 minutes. The efficacy data of vernakalant in converting AF to NSR in patients who had AF for longer than 7 days was not reported. Safety data for ACT 4 are detailed in the adverse events section below.
All three studies (ACT 1,3 and 4) demonstrated that vernakalant effective in converting acute AF to NSR when compared to placebo. Pooled post hoc analysis also showed similar efficacy of vernakalant in patients with or without ischemic heart disease, hypertension, and hepatic impairment; however patients with heart failure, renal impairment or those aged over 75 years achieved NSR less commonly. 29 However, the number of subjects in each of these subgroups is small and further studies are required to confirm these findings. So far, vernakalant has not been directly compared to other antiarrhythmic agents in terms of efficacy and tolerability. A phase III prospective, randomized, double-blind, active-controlled study is currently ongoing to evaluate the efficacy of IV vernakalant versus IV amiodarone in converting recent-onset AF (3 to 48 hours in duration) to sinus rhythm within 90 minutes after vernakalant administration. 30 Although efficacy comparisons between antiarrhythmics are useful, having amiodarone compared to vernakalant might not be the best choice, especially for the acute cardioversion of AF, due to prolonged infusions that are generally required for amiodarone's efficacy.
Post-Operative Atrial Fibrillation
ACT 2 is a Phase III double-blinded, randomized, placebo-controlled trial design to evaluate the efficacy and safety of IV vernakalant for the management of AF after valvular or coronary artery bypass surgery.27,28 One hundred and sixty-one patients who developed AF or atrial flutter between 3 hours and 7 days after surgery were randomized to receive IV vernakalant 3 mg/kg administered over 10 minutes or placebo. If AF or atrial flutter persisted after 15 minutes, another dose of vernakalant 2 mg/kg or placebo was given. Forty-seven percent of vernakalant treatment group responded as compared to 14% of placebo patients (p = 0.0002) in achieving NSR within 90 minutes of vernakalant administration. The median time to conversion to NSR was 12 minutes for the vernakalant group and 75% of those converted after receiving the first dose. Of the 10 patients with atrial flutter, no patients in the drug group and one patient in the placebo group converted to normal heart rhythm. However, the number of patients with atrial flutter is small and further studies are needed to evaluate the efficacy of vernakalant in patients with atrial flutter. The investigators concluded that vernakalant was effective in the conversion of atrial fibrillation to NSR after coronary bypass and valvular surgery. The use of other ventricular rate control agents, class I and III antiarrhythmic agents and anticoagulation was not described in full by the investigators.
Atrial Flutter
Efficacy of IV vernakalant in converting atrial flutter to NSR was evaluated in the Scene 2 study (unpublished data submitted to the FDA). 21 Only 1 out of the 39 patients in the vernakalant group (2.6%) had their atrial flutter converted to NSR within 90 minutes after initiation of treatment. No patient in the placebo group successfully converted to NSR. No further detail was provided regarding the exact demographics of the patient population and other concurrent therapies that were administered to these patients. The ACT 2 and 3 studies included a total of 33 patients with atrial flutter, but only 1 out of the 20 patients with atrial flutter in the vernakalant group converted to NSR.21,27,28 It was determined that vernakalant is not effective for the management of atrial flutter.
Maintenance of NSR
Preliminary efficacy of oral vernakalant in converting and maintaining NSR was initially explored in a phase 2a double-blind, placebo-controlled, randomized, dose-ranging study. 31 Patients who had experienced AF for more than 30 and less than 180 days were randomized to either vernakalant of 300 mg (n = 54), 600 mg (n = 56) or placebo (n = 49) twice daily. After the first 3 days, patients who are still in AF were electrically cardioverted. Successfully cardioverted patients continued to receive treatment for the remaining 25 days of the study. The results demonstrated a statistically significant difference between the 300 mg dosing group and the placebo group in terms of maintaining NSR at 25 days (61% vs. 43%; p = 0.048). For the 600 mg dosing group the difference trended toward, but did not reach statistical significance when compared to placebo (p = 0.060). When the two groups were combined, vernakalant was significantly superior to placebo (p = 0.028). This study provides early evidence for the efficacy of oral vernakalant in preventing the recurrence of AF. However, since the number of patients who received electrical cardioversion after the first 3 days of therapy was not reported, it is not known if oral vernakalant has demonstrated efficacy in converting AF to NSR in a group of patients who have relatively longer duration of AF compared to those enrolled in the studies evaluating the IV formulation. Larger study will have to continue to explore the efficacy and safety of the use of oral vernakalant.
In July of 2008 final results from a phase 2b double-blind, placebo-controlled, randomized, dose-ranging study were released by Cardiome Pharma Corporation. 31 This study was conducted to explore safety, and tolerability and efficacy of oral vernakalant in maintaining NSR over a 90-days period. A total of 735 patients were randomized in the study, but only 605 were successfully cardioverted to sinus rhythm and entered the 90-day maintenance phase during which oral vernakalant was evaluated for efficacy. Patients were randomized to receive 150 mg, 300 mg or 500 mg dose of vernakalant or placebo twice daily. The final analyses demonstrated statistical significant efficacy for the 500-mg dosing group of oral vernakalant as compared to placebo in maintaining NSR in day 90 (51% vs. 37%; p = 0.0221). A reduced rate of AF relapse was seen with both the 150 mg and 300 mg dosing groups, but the difference was not significant when compared to placebo. The median time to recurrence of AF was three times higher in patients receiving the 500 mg dosing of vernakalant versus placebo (>90 days vs. 27 days). This study demonstrated that oral vernakalant is safe and efficacious in maintaining NSR in successfully cardioverted patients.
Overall Efficacy
Based on the results of the clinical studies, overall, vernakalant when given as a 2 doses regimen (3 mg/kg + 2 mg/kg 15 minutes later if NSR is not restored) appears to be effective in converting subjects from short-term (onset 3 hours to 7 days) AF to NSR. The conversion rate on two doses was good (range 45%-63% conversion rate) and comparable to other agents reported with other acute conversion agent such as ibutilide (acute conversion rate 32%-51%), although no head to head comparison has been made between the two agents. 32 However, the strength of association between vernakalant and conversion to SR appeared to be reduced in subjects who failed to convert on the first dose. This led the FDA to believe that the population who failed to convert on one dose is to some extent resistant to the antiarrhythmic effect of vernakalant, and somewhat different from the population that responded on one dose. FDA recommended further analyses be performed to further explore the efficacy of vernakalant in these two patient populations, those who failed to convert on one doses versus those who did. 21
Safety
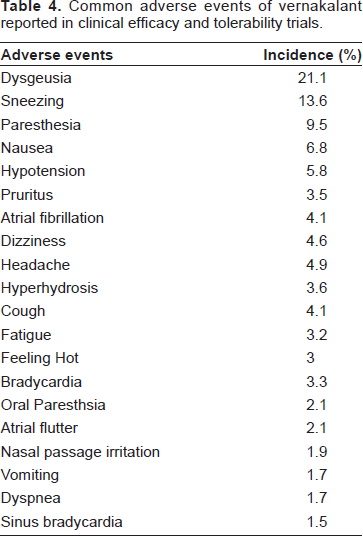
The vernakalant clinical trials program enrolled a total of 1198 subjects; 971 had AF and 86 had atrial flutter. Seven hundred and seventy-eight were exposed to vernakalant; and 585 subjects were included for short-term AF (onset between 3 hours and 7 days). Thirty six of the subjects who received vernakalant were healthy volunteers. The range of exposure was 0.1 mg/kg to 5 mg/kg with the majority receiving 2 to 5 mg/kg. Table 4 summarizes the overall incidence of common adverse events reported to the FDA.
Common adverse events of vernakalant reported in clinical efficacy and tolerability trials.
In the Phase II intravenous vernakalant study, 39 of the 56 patients experienced a total of 122 adverse events. 23 The incidence of side effects were similar among patients who received placebo, 0.5 mg/kg followed by 1 mg/kg dose of vernakalant, and 2 mg/kg followed by 3 mg/kg dose of vernakalant). The most commonly reported adverse events for vernakalant were cardiovascular related (7 [35%] in placebo, 4 [22.2%] in the 0.5 followed by 1 mg/kg group, and 3 [16.7%] in the 2 followed by 3 mg/kg group). The cardiac adverse events reported in the placebo group included 2 patients with nonsustained ventricular tachycardia, one patient with ventricular premature tachycardia and one with ventricular premature beats. An episode of transient cerebral ischemic attack occurred one day after conversion to NSR in one of the patients. Bradycardia and hypotension occurred in one patient, immediately after cardioversion and pulmonary edema in another patient, and recurrent AF in the another placebo patient. In the low dose vernakalant group, 2 patients experienced ventricular premature beats, one experienced sinus bradycardia and one experienced ventricular fibrillation, which was deemed to be due to an asynchronous discharge during an electrical cardioversion performed an hour after administrating the second infusion. In the high dose vernakalant group, 2 patients reported ventricular premature beats and one experienced sinus bradycardia.
In the Phase III clinical studies with IV vernakalant, during the 30 day follow-up period, 21 (18.3%) of patients in the placebo group and 29 (13.1%) in the vernakalant group experienced adverse events. Recurrent AF requiring hospitalization (17 in placebo and 22 in vernakalant group respectively) was the most commonly reported adverse event. No cases of TdP was reported. Other side effects of vernakalant reported include dysgeusia (66 [30%]), paresthesia (24 [11%]), sneezing (26 [16%]), nausea (20 [9%]), cough (11 [5%]), puritus (13 [6%]), and hypotension (14 [6%]).25–27
In ACT2, total adverse events were reported in 17(32%) and 41(38%) of patients receiving placebo and vernakalant, respectively, within the 24 hour period following study drug administration. The most common adverse events in patients given vernakalant were recurrent AF 21(20%), nausea 6(6%), constipation 5(5%), weight increase 5(5%) and dyspnea 5(5%). Rates of overall adverse events were similar to the placebo group (11% in placebo vs. 9% in the vernakalant group). Two patients (2%) given vernakalant experienced a serious adverse event (complete AV block and hypotension). 28
In terms of oral vernakalant, in the Phase IIa study, the drug appeared to be well-tolerated. 31 During the 28 days of dosing, adverse events that were related to the drug occurred in 1% of placebo patients, 4% of patients receiving 300 mg twice daily dosing, and 5% of patients receiving 600 mg twice daily dosing. There was no drug-related TdP reported. Details about the type of serious side effects are not available at this time. Safety of oral vernakalant, especially with long term use, will continue to be explored by the Phase IIb study.
Adverse events more commonly with vernakalant experienced, and at a higher incidence than placebo included sneezing, nausea, dysgeusia, paresthesia and hypotension. They were generally transient, and manageable in the setting for which vernakalant injection is intended, where patients are monitored by telemetry. Discontinuation rate was reported to be 3.8%. 21 Hypotension occurred in an overall incidence of 1.2% (9/773 patients) from all the studies with vernakalant (compared to 0.3% in placebo). 21 Sinus arrest, complete atrioventricular block and ventricular fibrillation, each occurred in 0.3% in patients receiving vernakalant in the study. 21 There was one (0.1%) event of an asymptomatic 9-beat run of TdP in the vernakalant group. However, this event occurred immediately after administration of ibutilide and was 2 hours and 20 minutes after vernakalant dosing was completed. 21 Vernakalant caused two cases of ventricular fibrillation, one of which was fatal, and they both occurred within minutes of the second dose. The incidence rate of ventricular fibrillation in this population was, probably, low because subjects with recent MI and advanced CHF were excluded. Although vernakalant does not affect ventricular myocardial repolarization significantly, it is not know how it will interact with substantially compromised myocardial tissue and function. Prolongation of the QRS complex and QT interval was observed after vernakalant injection, by 8 msec and 10 msec respectively. Despite the observed QT prolongation, incidence of ventricular arrhythmia was not different from the placebo group. The number of patients who had QTc intervals >550 msec was not significantly different between vernakalant and placebo groups. 21
Patient Preference
Management of atrial fibrillation involves achieving 3 major goals, ventricular rate control (rate control), preventing thromboembolic events and restoration of NSR (rhythm control). 2 Rate control involves the use of atrioventricular nodal blocking agents such as beta-blockers, calcium channel blockers or digoxin. Patients who receive rate control management strategy will require long-term anticoagulation with warfarin therapy to prevent thromboembolic events. Except if they only have one of the following risk factor: congestive heart failure, hypertension, age >75 years, diabetes and previous thromboembolic events, then aspirin may be used. Rhythm control involves the use of Class Ia, Ic and III antiarrhythmic agents such as procainamide, flecainide and amiodarone, for restoring and maintaining NSR. 2 Theoretically, once NSR is restored, anticoagulation therapy can be discontinued after a month. 2 However, in certain high risk patients, recent study evaluating rate versus rhythm control strategy indicated that long-term anticoagulation therapy may also be needed. 3 In recent years, numerous clinical trials have compared the rate control strategy versus rhythm control strategy in long term cardiovascular outcomes in patients with AF. From a therapy point of view, each strategy has its pros and cons. 4 Overall, rate and rhythm control are believed to produce non-significant differences in long-term cardiovascular outcomes. From the side effect profile perspective, beta-blockers, calcium channel blockers and digoxin are probably more manageable therapy to avoid TdP. However, patients who are on rate control therapy will have to be on warfarin therapy indefinitely. The numerous drug and food interactions with warfarin and the intensive laboratory monitoring with warfarin therapy probably significantly impact patient's life. In addition, the older the patient gets, the more difficult it is to tolerate warfarin therapy. Since their risk of bleeding from warfarin may increase due to their needs to be on more multiple medications, decrease in hepatic function with aging, the need to go through various surgical procedures for comorbid conditions …. etc. On the other hand, rhythm control therapy carries the risk of TdP, this risk increases in patients with other underlying cardiovascular diseases. Also patients may not have to deal with life-long warfarin therapy. In addition, in certain patient population group such as patients with severe heart failure, the improvement in “atrial kick” by restoration of NSR may improve symptoms and quality of life much better than the rate control strategy. 4 Therefore, the choice of therapy between rate versus rhythm control is as much depends on clinical data as patients’ preference. If one can improve the efficacy yet minimize the risk of Class Ia, Ic and III antiarrhythmic agents, that may make rhythm control a more attractive strategy to most than rate control strategy. With data available to-date, vernakalant appears to have low risk of TdP. However, the majority of the clinical data are with the intravenous form only that is used for acute cardioversion. Whether vernakalant can be the new promising rhythm control agent will have to depend on more data becoming available with the oral form for long-term use. The use of ventricular rate control strategy versus rhythm control strategy in AF continues to be controversial, whether vernakalant will change the strategy is not known at this point.
Place in Therapy
Clinical studies demonstrated that vernakalant is effective for conversion to NSR in patients with recent onset AF compared to placebo. Preliminary data also suggested that the oral vernakalant used continuously appeared to successfully maintain NSR vernakalant has atrial specific antiarrhythmic action. The incidence of TdP is low as observed in clinical studies. So far, there is one case of TdP reported in pre-marketing trials but the patient was also given ibutilide. However, none of the current clinical trial enrolled patients who are at higher risk of developing TdP and ventricular arrhythmias, such as those with decompensated heart failure, a population group that experience high incidences of AF. In addition, no studies described patients’ baseline AF symptoms. In the information submitted to the FDA, it was indicated that in all the studies combined, a statistically significant higher percentage of patients were symptom free in the IV vernakalant group compared with the placebo group in both the short-duration AF (3 hours to 7 days: 48.9% vs. 26.4%) and in the overall atrial fibrillation population (> 7 days: 46.3% vs. 28.0%) at 90 minute after drug administration, 21 In addition, it would be important to find out how many patients have duration of AF < 48 hours. Spontaneously conversion rate of NSR in these patients is high. 33 The use of antiarrhythmic agents for cardioversion may not be necessary in these patients and may only subject patients to un-necessary side effects. Furthermore, current data available do not describe in details the ventricular rate control strategy and anticoagulation management in patients with AF > 48 hours and any safety concern regarding thromboembolic events. In addition, head to head comparative trials with other antiarrhythmics and rate control agents are not available to-date, and will be needed to better define the role of vernakalant in AF management. If FDA makes their final approval of IV vernakalant today, it should be recommended to be used as an alternative agent for acute cardioversion only. Other agents available in the United States market, officially approved by the FDA for acute cardioversion include IV ibutilide. Other therapeutic alternatives (although not officially approved by the FDA for acute cardioversion), include IV procainamide and oral propafenone. Vernakalant has not been compared with any of these agents in terms of efficacy or tolerability.
Conclusions
Vernakalant is a new atrial-selective Class III antiarrhythmic agent. Phase III clinical studies of the IV formulation and preliminary data on the oral formulation indicate that vernakalant is efficacious in converting recent onset AF to NSR and maintaining NSR compared to placebo. The drug also appeared to be well-tolerated. Intravenous vernakalant is currently under FDA consideration for approval. The recommended dosage is 3 mg/kg infused over 10 minutes, followed by 2 mg/kg infused over 10 minutes if NSR is not restored 15 minutes later. Future studies need to further explore the comparative efficacy between vernakalant and other antiarrhythmic agents currently available, as well as the long-term tolerability with the oral formulation.
Disclosure
The authors report no conflicts of interest.