
Abstract
Colchicine is a substrate for cytochrome 3A4 (CYP3A4) enzyme and P-glycoprotein efflux transporter (P-gp); consequently, concomitant administration with drugs that inhibit these have the potential to cause clinically significant increases in colchicine plasma concentrations and precipitate adverse events. Ritonavir, a protease inhibitor, elicits potent CYP3A4 and P-gp inhibitory activity. In this open-label, nonrandomized, one-sequence, two-period study, 24 healthy volunteers received a single 0.6-mg dose of colchicine alone and together with multiple-dose ritonavir (100 mg twice daily for 4 days) to evaluate drug-drug interactions. Serial blood samples were collected for the determination of colchicine plasma concentrations. Standard pharmacokinetic parameter values were calculated along with 90% confidence intervals (ie, area under the concentration-time curve plasma from time zero to the time of last quantifiable concentration [AUC0-t and AUC0-∞], maximum drug concentration [Cmax]) for colchicine alone and colchicine combined with multiple-dose ritonavir. The mean Cmax and AUC0-t were significantly increased (170% and 245%, respectively) when colchicine was coadministered with ritonavir as compared with colchicine alone. Study data confirm the need for a dose adjustment (approximately 50% reduction) when colchicine is coadministered with strong CYP3A/P-gp inhibitors.
Introduction
Gout is a painful and progressive disease characterized by joint destruction and deformity, which has a substantial clinical and economic burden as evidenced by decreases in functionality, productivity, and quality of life.1,2 According to the National Health and Nutrition Examination Survey (NHANES), the estimated prevalence of gout among adults ≥ 20 years from 2007 to 2008 in the United States was 3.9% or 8.3 million individuals. 3 Colchicine is commonly indicated in this population for the treatment and prevention of gout flares and is generally well tolerated when used at recommended doses, although its therapeutic index is relatively narrow.4–6 An increased incidence of serious adverse events, including fatalities and life-threatening conditions, has been reported by the United States Food and Drug Administration (FDA) Adverse Event Reporting System database when colchicine is coadministered with certain P-glycoprotein efflux transporter (P-gp) or cytochrome 3A4 (CYP3A4) inhibitors. 7
Because CYP3A4 enzyme is the predominant drug-metabolizing enzyme in the CYP450 system, evaluation of the effect of drugs that influence this enzyme system is important. 8 Colchicine is primarily metabolized by CYP3A4, 9 and colchicine is also a substrate for P-gp.10,11
Protease inhibitors are often involved in clinically important drug interactions resulting from an alteration of cytochrome P450 metabolism. 12 Ritonavir was first approved in 1996 in the United States at the recommended dosage of 600 mg twice daily as part of a highly active antiretroviral therapy regimen. 13 Despite its potent antiviral activity, ritonavir was not well tolerated by human immunodeficiency virus (HIV)-infected patients when administered at the approved dosage and is therefore not approved as a single-dose agent for the treatment of patients with HIV. Instead, ritonavir is more routinely used as a pharmacokinetic enhancer or “booster agent” at lower than approved dosages and combined with other approved protease inhibitors that are CYP3A4 and P-gp substrates and have inherent poor bioavailability due to first-pass effect. The approved dosage of ritonavir when combined with other protease inhibitors is typically administered at 100 mg twice daily or 200 mg once daily. 14 An improvement in the overall bioavailability of coadministered protease inhibitors is due to the potent CYP3A4 and P-gp inhibitory activity of ritonavir.15–17 There are limited data on the prevalence of HIV and gout comorbidity although two separate studies have shown that asymptomatic hyperuricemia and gout are associated with ritonavir-boosted regimens in patients with HIV.18,19 Therefore, it is important to evaluate the effect of ritonavir on the pharmacokinetics of colchicine and to provide greater detail and further information on the magnitude of this drug-drug interaction than previously found in the literature. The FDA also required that the information be added to the labeling of all FDA-approved protease inhibitors. The objectives of the current study were to determine the effect of multiple-dose ritonavir on the pharmacokinetics of single-dose colchicine in healthy adults and to assess the safety and tolerability of single-dose colchicine administered alone and in combination with multiple-dose ritonavir.
Methods
Study design
This was an open-label, nonrandomized, single-center, one-sequence, two-period, drug-drug interaction study design. Following screening for eligibility, subjects received a single 0.6-mg dose of colchicine on day 1 under fasted conditions (Fig. 1). After a 14-day washout, subjects received ritonavir 100 mg twice daily (days 15–18), a dosage sufficient to achieve multiple-dose ritonavir concentrations. On the morning of day 19, subjects received a single dose of colchicine concomitantly with ritonavir 100 mg and then received a final 100-mg dose of ritonavir on the evening of day 19. Subjects were confined to the study unit on two separate occasions for 1.5 days each, beginning the afternoon of the day before the scheduled colchicine dose (day 1 and day 18), and remained confined until the morning after dosing occurred on day 2 and day 20 (ie, 24 hours postdose) during pharmacokinetic sample collection. Subjects were to return for nonconfined pharmacokinetic sample collection on days 2, 3, 4, and 5 (after the first single dose of colchicine) and days 19, 20, 21, and 22 (after the second single dose of colchicine). The study design is acceptable for evaluating the potential drug-drug interaction. The dose and regimen of each drug and the blood sample collection times were selected based on the pharmacokinetic properties of the two drugs administered. The study was conducted in accordance with the guidelines set forth by the International Conference on Harmonization Guidelines for Good Clinical Practice and the Declaration of Helsinki and was approved by the institutional review board of the study site. All subjects provided written, informed consent.
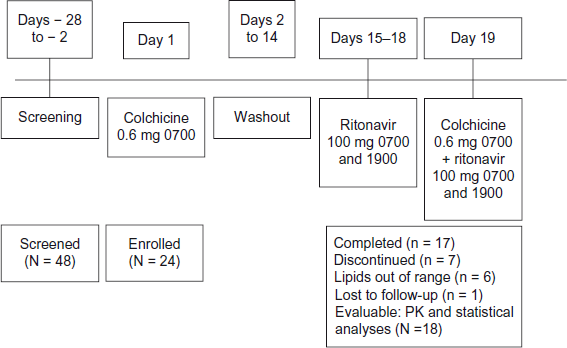
Study schematic and subject disposition.
Subjects
Twenty-four healthy, nonsmoking, adult volunteers (14 men and 10 women) aged 18 to 45 years with no risk factors for renal or hepatic impairment were enrolled in the study. Subjects were to have total cholesterol and triglyceride levels below the upper limit of normal and a body mass index ≥ 18 and ≤ 32 kg/m2 inclusive and a hemoglobin level ≥ 11.5 g/dL. Women of childbearing potential were to be either abstinent for 14 days before the first dose and throughout the study or using an acceptable method of birth control.
Exclusion criteria included a positive test for HIV, hepatitis B, or hepatitis C; a history/presence of a significant medical condition (ie, cardiovascular, pulmonary, hepatic, gallbladder/biliary tract, renal, hematologic, gastrointestinal, endocrine, immunologic, dermatologic, neurologic, or psychiatric); a recent (2-year) history of alcohol or drug abuse; and an active sexually transmitted disease.
Subjects were also not allowed to be on a special diet, to have participated in another clinical trial, to have donated blood within 28 to 56 days before the first dose, or to have used any drugs/substances known to inhibit or induce CYP450 enzymes and/or P-gp within 28 days before the first dose and throughout the study. Subjects were instructed to abstain from consuming prescription or over-the-counter medications, herbal products, or vitamins/supplements in suprapharmacologic doses within 28 days of study initiation. Subjects were also advised to avoid grapefruit-containing products within 14 days and caffeine and/or xanthine-containing products within 48 hours of study dosing.
Assessments
Serial blood samples to determine colchicine plasma levels were collected predose on days 1 and 19 and for 96 hours postdose (0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, and 24 hours [while confined] and 36, 48, 72, and 96 hours [nonconfined]). Blood samples were immediately placed on ice, centrifuged, and then frozen pending sample analysis. Colchicine plasma concentrations were analyzed using a validated liquid chromatography–-tandem mass spectrometry methodology performed at a central laboratory (PRACS Institute, Ltd., Cetero Research, Fargo, ND).
Standard pharmacokinetic parameter values were determined, including the following:
AUC0-t: area under the concentration-time curve from time zero to the time of the last quantifiable concentration (t)
AUC0-∞: AUC from time zero extrapolated to infinity
Cmax : maximum drug concentration
Tmax : time to Cmax
Kel: terminal elimination rate constant
Varea/F: apparent total volume of distribution
CL/F: apparent total body clearance
t1/2: elimination half-life
Safety was assessed by the observed and self-reported adverse events and via assessment of vital signs and laboratory parameters.
Pharmacokinetic analysis
Although a formal sample size calculation was not performed, a sample size of 24 was considered adequate based on sample sizes used in other published drug interaction studies with ritonavir. Pharmacokinetic parameter values could be estimated for 18 subjects who completed both study dosing regimens and for whom plasma concentration data were available. Data for the 6 subjects who did not receive colchicine in both dosing conditions were not included in the pharmacokinetic analyses but were included for any safety analyses. Data were included for subjects who did not complete the study but did complete a major portion of the study. Data from subjects with missing concentration values (eg, missed blood draws, lost samples, samples unable to be quantified) were to be used if pharmacokinetic parameters could be estimated using remaining data points; otherwise, data from these subjects were to be excluded from the final analysis.
Analyses of variance (ANOVA) were performed on the ln-transformed AUC0-t, AUC0-∞, and Cmax of colchicine with ritonavir (test) versus colchicine alone (reference) while Tmax was analyzed without transformation. The ANOVA model was to include treatment as a fixed effect and subject as a random effect. Each ANOVA included calculation of least-squares means (LSM), the difference between treatment LSM, and the standard error associated with the difference. These statistical analyses were performed using the appropriate SAS® procedure (SAS Institute, Inc., Cary, NC). Ninety percent confidence intervals (CIs) for each ratio were derived by exponentiation of the CIs obtained for the difference between treatment LSM resulting from the analyses on the ln-transformed AUC0-t, AUC0-∞, and Cmax. The CIs were expressed as a percentage relative to the reference regimen. If the 90% CI fell entirely within the range of 80% to 125%, a determination of no drug-drug interaction present was concluded. 8
Results
Disposition of study populations
Forty-eight subjects were screened for inclusion in this study. Eleven of these were screen failures, seven had a schedule conflict prior to period I check-in, and six were not needed for the study; therefore, a total of 24 subjects were enrolled. Because lipid panels were outside the normal range (not appreciated by study investigator before the first period conduct), 6 of the enrolled subjects discontinued before the second colchicine dosing period and were therefore not included in the pharmacokinetic analysis (Fig. 1). Demographic parameters are summarized in Table 1.
Demographics and baseline characteristics.
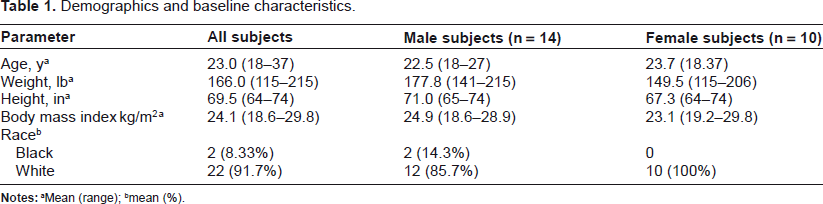
Mean (range);
mean (%).
Pharmacokinetics
Detectable colchicine concentrations were achieved for all subjects at the first postdose time point (30 minutes) and remained above the limit of quantitation in approximately 50% of all subjects within 12 hours postdose when dosed with colchicine alone and within 36 hours when given in combination with ritonavir (Fig. 2). Pharmacokinetic parameters for day 19 versus day 1 are summarized in Table 2. The Tmax of colchicine was not affected by concomitant administration of ritonavir, but Cmax was increased by 170% and AUC0-t was increased approximately 245%. This was accompanied by a decrease in total colchicine oral clearance of 70%. When pharmacokinetic parameters were evaluated by gender, exposure to colchicine was slightly higher in women compared with men. However, the difference appears to be due to differences in body weight because there was no difference between genders in weight-adjusted clearance.
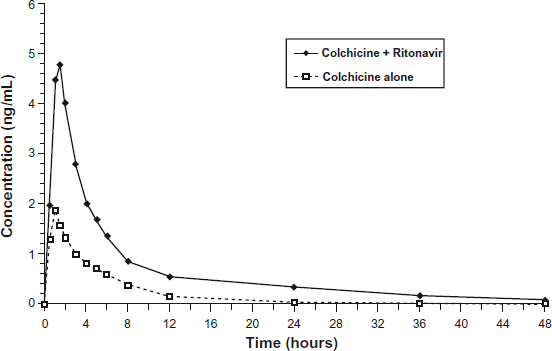
Mean colchicine plasma concentrations versus time profiles following administration of a single dose of colchicines 0.6 mg alone and with a 5-day regimen of ritonavir (N = 18).
Summary of colchicine pharmacokinetic parameters after administration of a single dose of colchicine 0.6 mg before and after a 5-day regimen of ritonavir (N = 18).
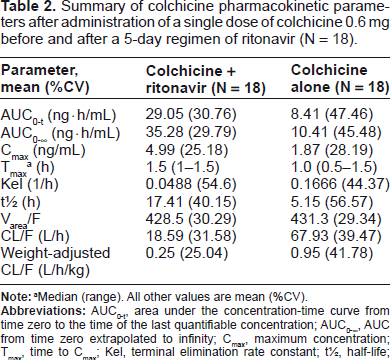
Median (range). All other values are mean (%CV).
Analyses performed on the ln-transformed pharmacokinetic parameters for day 19 versus day 1 are summarized in Table 3. The results indicate a significant drug-drug interaction between colchicine and ritonavir because the 90% CIs were outside the no-effect range (80%–125%) for the relevant pharmacokinetic parameters (AUC0-t, AUC0-∞, and Cmax).
Geometric means, ratio of means, and 90% CIs (N = 18).

Safety
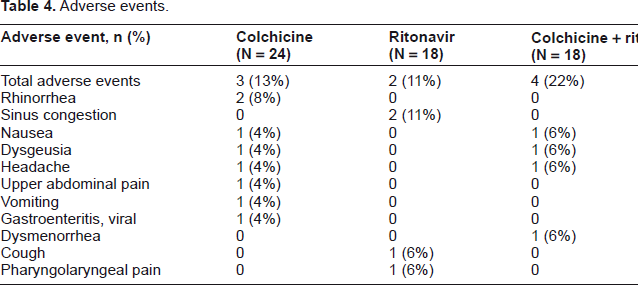
Five subjects (20.8%) experienced a total of 17 treatment-emergent adverse events during the course of the study. Nine adverse events occurred after treatment with colchicine alone, 4 occurred after treatment with ritonavir alone, and the remaining 4 occurred after coadministration of colchicine and ritonavir. The most common adverse events were nausea, rhinorrhea, and sinus congestion, each occurring in 2 subjects (8.3%, Table 4). There was no apparent difference in adverse events between the 2 dosing conditions despite the increase in exposure when given with ritonavir. All events were mild to moderate in severity, and none resulted in any subject being dropped from the study. There was also no significant effect on vital signs, and clinical laboratory values were considered unremarkable.
Adverse events.
Discussion
Colchicine is used for the treatment and prophylaxis of gout flares. The narrow therapeutic index of colchicine 20 suggests that any drug-drug interactions that significantly increase colchicine systemic exposures have the potential to increase the risk of colchicine-related toxicities. Assessing colchicine toxicity is not predictive, and pharmacokinetic analyses are necessary to understand drug interactions and determine appropriate dosing changes.
The current study shows that coadministration of colchicine with the potent CYP3A4 and P-gp inhibitor ritonavir significantly increases single-dose colchicine exposures in healthy volunteers. The study utilized a crossover design in the same subject and under the same study conditions, which is the methodology preferred by the FDA. 8 These results are consistent with other studies that have shown clinically significant drug-drug interactions among patients receiving concomitant strong CYP3A4/P-gp inhibitors such as clarithromycin, leading to elevated colchicine concentrations and the development of severe adverse events.21–24 In the current study, concomitant ritonavir resulted in increased colchicine exposures, as evidenced by increases in the mean colchicine AUC0-t and AUC0-∞ values of approximately 245% and 239% compared with administration of colchicine alone. Total apparent oral clearance was decreased by 70% when colchicine was coadministered with ritonavir as compared with the total apparent oral clearance when colchicine was administered alone (19 versus 68 L/h). These findings indicate a significant drug interaction occurs when single-dose colchicine and multiple-dose ritonavir are coadministered.
The significant interaction between colchicine and CYP3A4 enzyme and P-gp inhibitors has led to the development of a dose-reduction algorithm for colchicine when used in combination with such agents. 25 Concomitant use of a strong CYP3A4 and P-gp inhibitor such as ritonavir necessitates a colchicine dose reduction of approximately 50% for the acute treatment of gout or for the prophylaxis of gout flares. 25 Such adjustments will help to ensure that colchicine can be administered to patients with acute gout in a safe and effective manner.
Author Contributions
Conceived and designed the experiments: SW, JD, MWD. Analysed the data: SW, JD, MWD. Wrote the first draft of the manuscript: BF, JAS. Contributed to the writing of the manuscript: SW, JD, MWD, BF, JAS. Agree with manuscript results and conclusions: SW, JD, MWD, BF, JAS. Jointly developed the structure and arguments for the paper: SW, JD, MWD, BF, JAS. Made critical revisions and approved final version: SW, JD, MWD. All authors reviewed and approved of the final manuscript.
Funding
This study was sponsored by Mutual Pharmaceutical Company, Inc., which is now a part of the Takeda Pharmaceuticals USA Inc., family of companies, Deerfield, IL.
Competing Interests
MWD is employed by Mutual Pharmaceutical Company, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, and is the inventor of multiple patents pertaining to colchicine. JLD is employed by Salamandra, LLC, an independent consulting firm that provides strategic and technical advice to the pharmaceutical industry. SW is employed by Mutual Pharmaceutical Company, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. The funding source did not have influence on study design, data collection, analysis and interpretation of the data, and preparation or review of the report or the decision to submit for publication.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Footnotes
Acknowledgments
Medical editorial assistance was provided by Bret Fulton, RPh, and James A. Shiffer, RPh, Write On Time Medical Communications, LLC., and was funded by Mutual Pharmaceutical Company, Inc., a which is now a part of the Takeda Pharmaceuticals USA Inc., family of companies.
The authors would like to acknowledge Alexander Nasr, MPharm, PhD, and Thomas Lauterio, PhD (Mutual Pharmaceutical Company, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited) for their review and critical revisions for important intellectual content.
Data from this study were presented in part in an abstract and poster at the 14th International Workshop on Comorbidities and Adverse Drug Reactions in HIV, July 19–21, 2012, in Washington, DC.