
Abstract
Background
Selective and nonselective nonsteroidal anti-inflammatory drugs (NSAIDs) are indicated for the treatment of juvenile idiopathic arthritis (JIA). However, the effect of NSAIDs on blood pressure (BP) in children has not been rigorously examined.
Methods
In this randomized, double-blind, multicenter, active-controlled, 6-week trial, the safety and efficacy of celecoxib (50 mg twice daily [bid] or 100 mg bid) or naproxen (7.5 mg/kg bid) was evaluated in patients aged 2–17 years with JIA.
Results
The least squares (LS) mean difference (celecoxib – naproxen) in change from baseline to week 6/final visit in systolic BP was 1.10 (90% confidence interval, -0.56, 2.76). No significant LS mean differences in diastolic BP relative to baseline were reported. Treatment-emergent adverse events occurred in 48% of patients in each treatment group.
Conclusion
Both celecoxib and naproxen had no impact on BP, and both treatments had comparable safety profiles. Celecoxib, or naproxen, could be seen as suitable treatment options for pediatric patients with JIA.
Introduction
Hypertension is increasingly recognized in children as an important cause of morbidity,1–3 alongside other chronic conditions of childhood such as obesity and asthma. In children and adolescents, hypertension is defined as systolic blood pressure (SBP) and/or diastolic blood pressure (DBP) measurements ≥95th percentile for age, sex, and height, based on repeated measurements on at least three separate occasions. 4 The prevalence of pediatric hypertension is approximately 3%–5% in the United States5–8; however, this figure is expected to increase due to the close association between hypertension and obesity.2,4,8,9 Children and adolescents with high blood pressure (BP) are at risk of substantial long-term health risks including hypertension-associated target organ damage. 4
Hypertension is a well-recognized risk factor for cardiovascular and cerebrovascular disease. Both selective and nonselective nonsteroidal anti-inflammatory drugs (NSAIDs) may exacerbate underlying hypertension in adults, particularly in patients who are receiving antihypertensive therapies such as angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, diuretics, or beta-blockers. 10 Therefore, all prescription NSAIDs, including selective and nonselective NSAIDs, carry the same warning for cardiovascular events from the US Food and Drug Administration. 11 However, based on a meta-analysis of adult arthritis trials, there is no evidence for increased risk of cardiorenal adverse events (AEs) or effects on BP with celecoxib, a cyclooxygenase-2 (COX-2)–selective NSAID, compared with nonselective NSAIDs and specifically naproxen. 12 Few trials have evaluated the impact of chronic NSAID therapy on BP in children. In a multicenter, randomized, double-blind, noninferiority trial, no apparent differences in BP were observed between children who were randomized to receive celecoxib or naproxen for a period of 12 weeks. 13 This was the pivotal efficacy, safety, and pharmacokinetic study that supported the Food and Drug Administration approval of the juvenile idiopathic arthritis (JIA) indication for celecoxib in the United States. However, the methodology used to collect the BP measurements in that study was not as rigorous as the methodology for studies in which BP is the primary endpoint.
The present study evaluated the effect of 6-weeks’ NSAID treatment on BP in patients with JIA. The primary objective was to compare the changes in SBP when pediatric patients were treated with celecoxib or naproxen.
Methods
Study Design
This study was a randomized, double-blind, multicenter trial in patients with JIA (oligoarticular, polyarticular arthritis, and children with systemic onset disease, but inactive systemic features). Children who were patients of a pediatric rheumatology specialist at participating centers and also met enrollment criteria were invited to participate in the study. Each patient was given a computer-generated randomization number, which was accessed by an interactive voice response system, to enable assignment to one of the drugs by the study personnel. Patients were randomized (1:1 ratio) to receive either celecoxib (capsules, 50 mg twice daily (bid) for patients weighing ≥10 kg and ≤25 kg or 100 mg bid for patients weighing >25 kg) or naproxen (suspension, 7.5 mg/kg bid, maximum dose of 500 mg bid) for 6 weeks (Fig. 1). The volume/dose of the study medications was determined according to the patient's weight at the baseline visit. There were five study visits, during which vital signs, BP measured by auscultation in triplicate (a fourth measurement of BP could be taken if a result was considered to be an outlier), and concomitant medication use were assessed. Physical examination and clinical laboratory findings were assessed at visit 1 and visit 5, with Tanner stage of development assessed at visit 1 only. Patient's and/or parent's global assessment of overall well-being was assessed at baseline and week 6. This assessment consisted of the parent/legal guardian of the patient placing a vertical line on a visual analog scale of 1–100 mm (where 0 = very well and 100 = very poor) to indicate a response to the statement: “Considering all the ways that arthritis affects your child, rate how your child is doing”. Patients aged ≥8 years at baseline also evaluated their own well-being using the same method, in response to the statement “Considering all the ways that arthritis affects you, rate how you are doing”. AEs were evaluated during visits 2–5. Patients who enrolled in the exploratory 24-hour ambulatory BP monitoring (ABPM) substudy had ambulatory BP measurements taken in addition to clinic BP measurements obtained using auscultation. ABPM measurements in a subgroup of participants were assessed at visits 1 and 5.
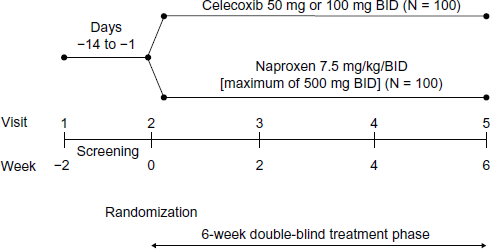
Study design.
Ethics Committee approval was obtained by each of the 32 centers at which the study was conducted. The institutional review board and/or an independent ethics committee at each center approved the protocol. The study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation guideline on Good Clinical Practice, and local regulatory requirements and laws. The patient's parent or legal guardian provided written informed consent prior to enrollment.
Study Eligibility
Eligible patients were aged 2–17 years with a body weight ≥10 kg at the baseline visit and had poly-articular (both rheumatoid factor positive and rheumatoid factor negative), oligoarticular (both persistent and extended types), and extended JIA for ≥3 months meeting the International League of Associations for Rheumatology criteria for JIA. 14 Patients must also have been considered candidates for chronic NSAID therapy to be included in the study. Patients with systemic JIA with active arthritis in at least 1 joint but without active systemic features were also considered for inclusion. Patients with psoriatic arthritis, enthesitis-related arthritis, and undifferentiated arthritis or with systemic JIA with active systemic features were excluded. Females of child-bearing potential (defined as menarche or ≥10 years of age, whichever occurred sooner) were required to use adequate contraception (including abstinence if the investigator deemed appropriate) and to have a negative urine pregnancy test prior to administration of study medication.
Exclusion criteria included (1) psoriatic arthritis, enthesitis-related arthritis, and undifferentiated arthritis types of JIA; (2) active systemic features over the prior 12 weeks in children with systemic JIA; (3) use of current NSAID or salicylate compounds, anticoagulants, lithium, cyclosporine, tacrolimus (FK 506), antihypertensives, any medication that in the investigator's judgment was expected to affect BP, and any medication within 30 days prior to screening; (4) initiation of or the change in dose of disease-modifying antirheumatic drugs and/or biologics within 30 days of screening; (5) oral or injectable corticosteroids administered within 2 weeks of screening/baseline; (6) known hypersensitivity allergy to sulfonamides, COX-2-selective inhibitors, aspirin, or NSAIDs; (7) diagnosis or treatment for esophageal, gastric, pyloric channel, or duodenal ulceration within 60 days of screening; (8) active gastrointestinal disease, a chronic or acute renal, or hepatic disorder, a significant coagulation defect, or any condition that in the investigator's opinion might preclude the use of an NSAID; (9) aspartate transaminase/serum glutamic oxaloacetic transaminase, alanine transaminase/serum glutamic pyruvate transaminase, creatinine, or blood urea nitrogen >1.5x the upper limit of normal for age/sex-adjusted normal values per the central laboratory or any other laboratory abnormality considered to be clinically significant within 14 days prior to baseline; (10) hypertension defined as SBP and/or DBP values ≥95th percentile for age, sex, and height for three measures at baseline visit; (11) active malignancy of any type or history of malignancy; (12) any significant, uncontrolled chronic condition or any other condition that would contraindicate study participation or confound interpretation of the results; (13) plans for surgical intervention during the study; (14) previous participation in this study; and (15) unlikely compliance with study procedures, including calm participation in BP assessments.
Safety Endpoints
The primary safety endpoint was the change from baseline to week 6/final visit for SBP.
Secondary safety endpoints were the change from baseline to weeks 2 and 4 for SBP and the change from baseline to weeks 2, 4, and 6/final visit for DBP. Other safety measures were overall safety and tolerability, which was evaluated by clinical laboratory measurements (serum chemistry, hematology, and urinalysis), physical examination, assessment of vital signs, and monitoring of the frequency and severity of AEs.
Efficacy Endpoints
Efficacy endpoints included (1) change from baseline to week 6/final visit in parent's assessment of overall well-being; (2) the number of patients at week 6/final visit with ≥30% improvement in the parent's global assessment of overall well-being; (3) change from baseline to week 6/final visit in the patient's assessment of overall well-being; and (4) the number of patients at week 6/final visit with ≥30% improvement in the patient's global assessment of overall well-being.
BP Assessments
At each visit, BP was measured by auscultation. After a 15-minute rest period, BP was measured with an appropriately sized BP cuff with the child in a seated position, with the back supported and feet flat on the floor. Three BP measurements were obtained at an interval of 5 minutes. 4 At week 6/final visit, BP was measured at trough (prior to the morning dose of study medication at site) and at peak exposure (approximately 2 hours after the morning dose). These two measurements were compared with those obtained at baseline and captured as peak trough measurements. Prior to study initiation, investigators at each site were trained in BP measurement methodology according to current guidelines for children. 4
24-Hour ABPM
A total of 24 patients were also enrolled in the exploratory 24-hour ABPM substudy. These patients had ABPM measurements obtained in addition to BP measurement using the cuff technique. Cuff BP measurements were to be taken first. Measurements were obtained using standardized equipment (Space Labs 90207 monitor) and a central ABPM reading laboratory performed data collection, reading, quality evaluation, and data transmission. Families received detailed instructions on the use of the monitor, which was programmed to inflate and record BP at prespecified intervals every 20 minutes from 06:00 to 20:00 and every 30 minutes from 20:01 to 05:59.
ABPM parameters for analysis included 24-hour average SBP, DBP, and heart rate; awake average SBP, DBP, and heart rate; and asleep average SBP, DBP, and heart rate.
Statistical Methods
BP change from baseline was analyzed using analysis of covariance with baseline height, weight, age, and BP as covariates. The least squares (LS) mean change from baseline and 90% or 95% confidence intervals (CIs) were generated. Change from baseline BP was also analyzed using a repeated measures model for sensitivity analysis. All randomized patients who received at least one dose of study medication were eligible for analysis. The primary analysis was conducted using the safety population. Sample size calculation was based on the primary efficacy safety measure (change from baseline to week 6/early termination in SBP). A sample size of 100 patients per treatment group would provide a 90% CI with an expected width of 4.42 mm Hg. The standard deviation (SD) used for the sample size calculation was based on a recent trial conducted by Pfizer in JIA patients (Study N49–01–02–195), which yielded a SD of 9.5 mm Hg for change in SBP. With 100 patients per group and with the above assumption on the SD, the study had 90% power to detect 3.95 mm Hg difference between the celecoxib and naproxen treatment groups, at the α-level of 0.1. Similarly, 100 patients per group with the same SD assumption had 90% power to detect 4.42 mm Hg difference between the celecoxib and naproxen treatment groups, at the α-level of 0.05. Except for the purposes of determining the sample size, no other comparisons between the present study and Study N49-01-02-195 13 were drawn.
Results
Patients
This trial was conducted at 32 centers, across 10 countries (Chile, Costa Rica, Peru, Philippines, Russian Federation, Serbia, South Africa, Switzerland, Ukraine, and United States). A total of 201 patients were randomized (Fig. 2). Five patients in the celecoxib group discontinued due to AEs, including three attributed to worsening of arthritis, one to headache, and one to urticaria/hives. Mean age was 11.1 and 11.2 years in the celecoxib and naproxen groups, respectively. The distribution of patients according to age groups was ≥2 to <8 years, n = 22 and 18; ≥8 to <13 years, n = 34 and 47; and ≥13 to <18 years, n = 44 and 33, for celecoxib and naproxen, respectively. Tanner stage assessments of development were similar between treatment groups. The median duration from the first diagnosis was 2.7 and 3.1 years, respectively, for celecoxib and naproxen. The median duration of treatment in both groups was 43 days and median compliance rate was 97.6% for celecoxib and 97.1% for naproxen.

Patient disposition.
Safety Endpoints
Change from baseline to week 6/final visit in SBP
The mean (SD) SBP at baseline was 98.5 (9.49) and 98.1 (9.49) mm Hg, and at week 6/final visit was 98.9 (8.84) and 97.4 (10.33) mm Hg for celecoxib and naproxen, respectively Table 1. The LS mean difference (celecoxib – naproxen) was 1.10 (1.004) with a 90% CI of -0.56, 2.76.
Change from baseline to week 6/final visit in SBP (safety population).

Change from Baseline to Weeks 2 and 4 in SBP
The LS mean differences of SBP at weeks 2 and 4 relative to baseline between celecoxib versus naproxen treatment were 1.09 (95% CI, -0.39, 2.57; P = 0.148) and 1.84 (95% CI, 0.21, 3.46; P = 0.027), respectively, indicating somewhat lower SBP in the naproxen treatment arm compared with no change in the celecoxib treatment arm.
Change from Baseline to Weeks 2, 4, and 6/Final Visit in DBP
The mean (SD) DBP at baseline was 62.2 (7.82) and 62.0 (7.61) mm Hg, and at week 6/final visit 61.8 (6.78) and 61.5 (7.64) mm Hg for celecoxib and naproxen, respectively. The LS mean differences of DBP at weeks 2, 4, and 6/final visit relative to baseline between celecoxib versus naproxen treatment were -1.21 (95% CI, -2.67, 0.26; P = 0.106) at week 2, -0.22 (95% CI, -1.30, 1.74; P = 0.776) at week 4, and -0.18 (95% CI, -1.69, 1.33; P = 0.815) at week 6/final visit.
Ambulatory BP Monitoring
ABPM measurements were analyzed for 23 subjects in total; 12 were randomized to celecoxib and 11 subjects were randomized to naproxen. After 6 weeks of treatment, subjects in the celecoxib group experienced an increase of the 24-hour mean ± SD SBP of 2.4 ± 7.0 mm Hg while subjects in the naproxen group experienced a decrease of 1.7 ± 12.4 mm Hg. Further review of the ABPM data identified an outlier in the naproxen group, whose baseline BP values were higher than the mean values for the group and clinically implausible. A post hoc sensitivity analysis, conducted without this subject, showed an increase of the ABPM 24-hour mean ± SD SBP by 1.9 ± 4.1 mm Hg.
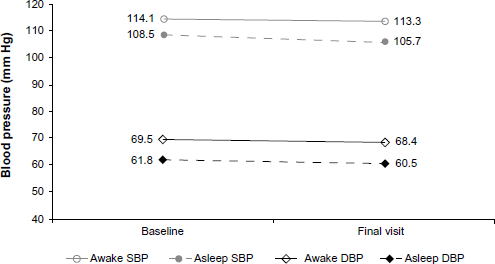
SBP and DBP changes from baseline to final visit in mean awake and asleep values are shown in Figures Fig. 3, Fig. 4, and Fig. 5. In the celecoxib group, the mean awake values increased by 3.1 mm Hg and the asleep values by 1.7 mm Hg. In the naproxen treatment group, the awake values decreased by 0.65 mm Hg and the asleep values by 3.2 mm Hg. After excluding subject 10031002 (outlier), the changes became similar to the celecoxib group: an increase of 2.3 mm Hg for the awake mean SPB value and of 1.2 for the asleep mean values. DBP values followed a similar pattern.

Mean SBP and DBP awake and asleep values at baseline and final visit (celecoxib).
Mean SBP and DBP awake and asleep values at baseline and final visit (naproxen).

Mean SBP and DBP awake and asleep values at baseline and final visit (naproxen, excluding subject 10031002).
Adverse Events
The proportions of treatment-emergent adverse events (TEAEs) were similar for patients in both treatment groups, with 48 (48.0%) patients in the celecoxib group reporting 82 events, and 47 (48.0%) patients in the naproxen group reporting 83 events. The proportion of patients reporting treatment-related TEAEs was slightly lower in the celecoxib group (13.0%) compared with the naproxen group (20.4%). An overview of TEAEs reported by ≥2% of patients during the double-blind treatment phase is provided in Table 2. Commonly reported all-causality TEAEs (incidence of ≥5% in any treatment group) included nausea, headache, and arthralgia. For nausea in both treatment groups and headache in the naproxen group, the majority of events were considered treatment related. No patients died in this study; one serious AE was reported in the naproxen group (a hand fracture) that was not related to the study treatment.
Summary of TEAEs and incidence of events occurring in ≥2% patients in any treatment group (safety population).

There were no relevant changes from baseline in values of any clinical laboratory, coagulation, urinalysis, or vital sign variable for either treatment group.
Efficacy Endpoints
Global Assessment of Overall Well-Being
Parents/guardians completed the assessment of overall well-being for all study patients, except for those ≥8 years of age, who completed their own assessments. The difference of the LS mean changes in the overall well-being from baseline to week 6/final visit between celecoxib versus naproxen treatment was 3.03 mm (95% CI, -2.76, 8.82; P = 0.303) according to the parent/guardian's assessment and -0.40 mm (95% CI, -6.50, 5.69; P = 0.897) according to the patient's assessment. According to parents’ assessments, 49% and 55% of the patients in the celecoxib and naproxen groups, respectively, showed ≥30% improvement in overall well-being. According to patients’ assessments, 44% versus 54% in the celecoxib and naproxen groups, respectively, showed ≥30% improvement in overall well-being. There were no significant differences in either parent/guardian- or patient-reported overall well-being between treatment groups.
Discussion
This study was carried out to examine the effect of chronic NSAID therapy on BP in patients aged 2–17 years with JIA. After 6 weeks of treatment with celecoxib or naproxen, no significant changes in SBP from baseline were observed with either NSAID. The LS mean difference (celecoxib – naproxen) was 1.10 (1.004) with a 90% CI of -0.56, 2.76. Therefore, it can be interpreted with 90% confidence that there was no difference in SBP changes from baseline to week 6/final visit between the celecoxib and naproxen groups.
These findings are consistent with those from a previous study 13 that compared the effects of celecoxib and naproxen in patients with JIA. In a post hoc analysis of BP data from that study, there was no difference between the two treatments in SBP or DBP change from baseline. In this study, we obtained similar results (with 95% confidence) and detected no significant difference when changes in SBP from baseline measurements were assessed at week 2 and for DBP at weeks 2, 4, and 6/final visit. The evaluation of the changes in SBP at week 4 indicated a treatment difference between celecoxib and naproxen, but this difference was small and not considered to be clinically important. The results of the sensitivity analysis, which applied a repeated-measures model for weeks 2, 4, and 6 were consistent with the primary analysis.
A retrospective cohort study in adults, which compared the effects of celecoxib, naproxen, and ibuprofen on BP in adult patients with hypertension, demonstrated that all three NSAIDs were associated with moderate mean increases (2 mm Hg) in SBP. 10 Consistent with these data and with the known vascular and hemodynamic effects of NSAIDs, our assessment of 24-hour ABPM measurements showed that both celecoxib and naproxen increased 24-hour SBP and DBP slightly. After 6 weeks of treatment, both celecoxib and naproxen exhibited nominal changes in 24-hour mean SBP: celecoxib increased mean (SD) SBP by 2.4 ± 7.0 mm Hg while naproxen increased SBP by 1.9 ± 4.1 mm Hg after exclusion of an outlier from the analysis, whose data were considered to be clinically implausible. These slight increases were similar for both drugs and were not considered to be clinically relevant. Awake and asleep mean values for SBP and DBP followed a similar pattern.
This study also evaluated efficacy of the study treatments via the patient's and parent's assessments of overall well-being. No statistically significant differences in the LS mean changes from baseline to week 6/final visit were observed between celecoxib and naproxen for these assessments, and a similar proportion of patients in each treatment group) reported ≥30% improvement in overall well-being. The efficacy of celecoxib for the treatment of JIA has previously been confirmed in a randomized, double-blind, multicenter study that assessed the effects of 3 mg/kg or 6 mg/kg doses with 7.5 mg/kg naproxen. 13 When the effect of celecoxib treatment was assessed against the American College of Rheumatology's pediatric 30% improvement criteria, 15 both doses were found to be at least as effective as naproxen.
In this study population, celecoxib and naproxen demonstrated comparable and acceptable safety and tolerability profiles that were consistent with their known profiles. Most TEAEs were considered to be of mild to moderate intensity. A limitation of this study is the relatively short duration of the observation period (6 weeks). Furthermore, ABPM data were collected from only a small subset of patients, making it difficult to extrapolate these results. Nevertheless, from the results of this study, it can be concluded that celecoxib is a safe and well-tolerated treatment option for pediatric patients.
Summary
The main purpose of this study was to evaluate the treatment effect of celecoxib, compared to naproxen, on blood pressure (BP) in pediatric subjects with juvenile idiopathic arthritis (JIA). Celecoxib is approved by the US Food and Drug Administration for the treatment for the JIA; however, there are limited data about the cardiovascular effects of nonsteroidal anti-inflammatory drugs in children. This study examined the effect of celecoxib treatment on BP levels in a pediatric population. The results demonstrate no significant effect on BP of either celecoxib or naproxen in children. These findings can support physicians’ decision-making when prescribing safe and effective treatments for children and young people who have JIA.
Author Contributions
Conceived and designed the experiments: BF. Ensured training of all study sites on uniform protocol for BP measurement in children: BF. Analyzed the data: MB, PB, DI. Wrote the first draft of the manuscript: MB, PB, DI. Contributed to the writing of the manuscript: BF, RN, LZ. Agreed with manuscript results and conclusions: MB, BF, RN, LZ. Jointly developed the structure and arguments for the paper: BF. Made critical revisions and approved final version: BF, RN. All authors reviewed and approved the final manuscript.
Footnotes
Acknowledgment
Editorial support was provided by Kate Bradford, PhD, of PAREXEL. This study was sponsored by Pfizer Inc.