
Abstract
We report the case of a 37-year old man presenting with a left ventricular cardiac thrombus in the setting of subclinical paroxysmal nocturnal hemoglobinura (PNH) developing two years after immunosuppressive therapy for thymoma-associated aplastic anemia. The literature regarding the interplay between autoimmunity and immunosuppression, aplastic anemia, thymoma and the emergence of PNH is reviewed.
Case Report
A previously healthy, 37-year old man presented initially in December 2004 with epistaxis, petechial rash and severe pancytopenia (absolute neutrophil count ANC 400/μL, hemoglobin 7.7 g/dL, platelets 3 × 103/μL). Bone marrow biopsy demonstrated severely hypocellular marrow with polyclonal lymphoid aggregates and normal karyotype, consistent with severe aplastic anemia (Figure 1). Viral serologies, as well as CD55/59 flow cytometry screening were negative. A CT scan performed during the initial workup revealed an 8 cm lobulated anterior mediastinal mass. The patient underwent a biopsy and subsequent thoracotomy, which revealed type B (cortical epithelioid) thymoma with evidence of the innominate vein invasion, infiltration of mediastinal fat and positive surgical margin, prompting subsequent adjuvant radiation to the surgical bed. Despite treatment of thymoma, there was no observed hematologic improvement in the aplastic anemia with continued, heavy transfusion dependence. The patient was evaluated for allogeneic bone marrow transplantation, but he did not have an eligible sibling and declined the option of a matched unrelated donor transplantation. He was treated with the standard course of antithymocyte globulin (ATG) and cyclosporine A without any evidence of hematologic response during the subsequent 6 months of support with nearly weekly platelet and/or red cell transfusions. However, over the course of the ensuing year his transfusion requirements spontaneously decreased and he ultimately achieved a partial remission from aplastic anemia. Since December 2006 he did not require any further transfusions.
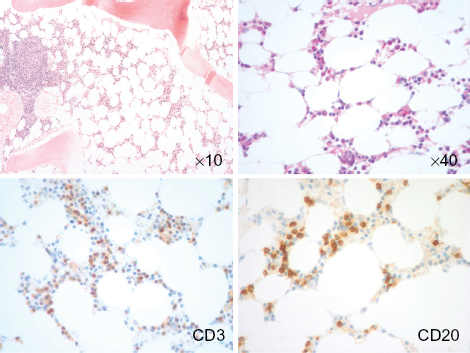
Microscopic images of the bone marrow biopsy. (
In November 2007, a routine surveillance CT scan of the chest incidentally showed a filling defect within the apex of the left cardiac ventricle, suspicious for a malignancy or thrombosis. Further investigation with a 2D echocardiogram, cardiac catheterization and MRI (Figure 2) confirmed a 3.4 cm tethered, mobile intraventricular mass and no cardiac function abnormality. The patient was anticoagulated with warfarin, however after 2 months there was no resolution of the lesion and excision was recommended.

Images of the cardiac magnetic resonance imaging (MRI) and 2D echocardiogram sequence visualizing the intraventricular thrombus. (
Considering prior median sternotomy, the mass was removed by minimally invasive thoracotomy under cardiopulmonary bypass. Pericardium was accessed through a small right lateral thoracotomy in the third intercostal space, with exposure of the interatrial septum above the pulmonary veins and dissection of paraaortic adhesions. Antegrade cardioplegia was delivered after heparinization. The mass was accessed via left atrium with retraction of the anterior leaflet of the mitral valve. It appeared as a yellowish fibrous structure attached to the trabeculations in the apex. The lesion was pathologically identified as organizing thrombus with peripheral fibroblast proliferation and fibrin inflammatory debris. There was no evidence of antiphospholipid antibodies, other thrombophilias, abnormal cardiac function or coronary vessel pathology. However, a repeat CD55/59 flow cytometry confirmed a new diagnosis of paroxysmal nocturnal hemoglobinuria (PNH), with over 80% CD55/59-deficient red cells and granulocytes (Figure 3). Continued anticoagulation was prescribed.

CD59 red cell flow cytometry histogram at the initial presentation and at the time of thrombus diagnosis, when a bimodal distribution with a dominant PNH clone became manifest.
At that point there was only mild chronic hemolysis (hemoglobin 11 g/dL, LDH 380 IU/L) and the patient did not require any intervention except for continued anticoagulation and monitoring. However, in June 2011 he sustained an episode of severe hemolysis (with a hemoglobin drop from 13.2 to 6.3 g/dL) following an upper respiratory tract infection, complicated by acute renal failure. Upon recovery, he was reevaluated for potential allogeneic bone marrow transplantation and eculizumab therapy. Figure 4 tracks the history of the patient's cytopenias on the background of clinical events.

Tracing of the patient's cytopenias (platelets—green, haemoglobin—red, neutrophil count—blue) on the backdrop of the clinical events.
Discussion
Paroxysmal nocturnal hemoglobinuria is a rare clonal stem cell disorder, characterized by chronic intravascular hemolysis, highly thrombophilic state and progression to bone marrow failure, commonly after a period of sustained hematopoiesis. 1 The pathogenesis of this disorder involves a somatic mutation in the PIG-A gene with consequent deficiency in glycosylphosphatidylinositol(GPI)-anchored membrane proteins, such as surface complement deactivators CD55 (decay-accelerating factor) and CD59 (protectin). This deficit leads to complement-mediated red cells lysis, increased platelet reactivity, abnormal fibrinolysis and a number of other pathophysiologic phenomena producing a hypercoagulable state. 2 Intravascular hemolysis with release of free plasma hemoglobin contributes to nitric oxide deficiency and endothelial dysfunction in a mechanism that links thrombotic risk in PNH and other hemolytic disorders such as sickle cell disease. 3
Thrombosis, the major source of morbidity and mortality in PNH, typically occurs within the venous circulation, frequently in unusual sites, such as hepatic or splanchnic veins, cavernous sinus etc. The 10 year cumulative incidence of venous thrombosis among all affected patients is 23% and depends on the size of the abnormal clone. Intraventricular cardiac thrombi have not been previously reported in association with this disease. Thrombosis within the left ventricle occurs almost exclusively in the setting of pre-existing heart failure. In a recent review only seven prior cases of left ventricular thrombus with preserved cardiac function were reported, mostly with a history of prothrombotic medical conditions. 4
Arterial thrombosis occasionally does occur in PNH. Among nine patients with acute stroke and PNH, in five cases the thrombosis was one of the first manifestations of the hematologic disorder. 5 In the largest descriptive series, cases of arterial thrombosis in the central nervous system, coronary circulation, hepatic, mesenteric arteries and aorta were summarized. 6 The arterial events were classified as high-risk despite their relatively low incidence. They occurred mostly in young patients without underlying atherosclerosis. However, the relative risk of coronary event was over 20 and cardiac thrombosis was associated with the highest mortality score.
The disturbed coagulation system in low-shear, slow-flow venous circulation is believed to play the dominant role in the pathogenesis of venous thromboembolism, while platelet pathology is typically implicated in arterial thrombosis. Interestingly, platelet activation was reported in PNH and antiplatelet therapy was suggested as possible therapeutic intervention. 7 Platelets just as other blood cells originate from the pathologic clone and are prone to complement-mediated injury and activation. Circulating platelet-derived procoagulant microparticles rich in phospholipids were detected in PNH and may contribute to the thrombotic risk. 8 In case of either venous or arterial thrombosis, anticoagulation remains the mainstay of therapy. Recent reports highlight decrease in thrombotic risk after treatment with eculizumab (monoclonal antibody against the complement protein 5). 9
The occurrence of contemporaneous malignant thymoma and severe aplastic anemia with subsequent PNH in our case sheds additional light on the complex interaction between thymoma-derived autoimmunity, T-cell directed immunosuppression and the dynamics of PNH clone emergence and survival. Sensitive assays detect cells with PNH phenotype in over 50% of patients with acquired AA, and the emergence of a PNH clone in AA had been previously reported in 10%–25% of cases after immunosuppressive therapy. 10 In the era of sensitive flow cytometry screening, although there is a large proportion of PNH-positive patients at the onset of AA, the occurrence of a new clone years later is not common. 11 In the majority of patients with a detectable mutant population, the proportion of PNH cells ultimately decreased after immunosuppressive therapy. 12
Thymoma often produces paraneoplastic autoimmune disorders, such as myasthenia gravis, pure red cell aplasia and AA. In the single previously reported case of PNH occurring after resection of thymoma, circulating myelotoxic CD8+ lymphocytes were detected. 13 It is hypothesized that while PIG-A mutations occur sporadically in healthy population, impaired immune surveillance or autoattack against normal progenitors is critical for expansion and hematopoietic dominance of the PNH clone. The mutant population has the ability to restore hematopoiesis—in our patient ironically leading to remission from severe aplastic anemia unresponsive to standard treatment. Such a paradoxical mechanism had been conjectured in the past. 14
Yearly screening for PNH in AA patients after therapy has been recommended by the International PNH Interest Group, although the benefits and duration of such screening remain uncertain. 15 While prospective series do not uphold earlier postulations that overt PNH should frequently develop in patients with AA harboring a small CD55/59 deficient population, our case underscores the importance of longitudinal follow up and screening. 16 The appearance of a new large PNH clone, while uncommon, warrants prophylactic anticoagulation, since the risk of thrombosis approaches 50% over 10 years. 17 Cardiac surgery in the setting of PNH may be complicated with increased risk of hemolysis, renal failure and thrombosis, so additional perioperative interventions have been recommended. 18 In case of clinically overt PNH, treatment with eculizumab is effective in decreasing hemolysis, transfusion dependence and the risk of thrombosis. 19 It also favorably affects survival in patients with PNH. 20 As our case illustrates, a seemingly spontaneous, delayed remission from AA may occur thanks to the emergence of hematologically effective PNH, which nevertheless can manifest its own thrombotic and hemolytic consequences.
Disclosures
Author(s) have provided signed confirmations to the publisher of their compliance with all applicable legal and ethical obligations in respect to declaration of conflicts of interest, funding, authorship and contributorship, and compliance with ethical requirements in respect to treatment of human and animal test subjects. If this article contains identifiable human subject(s) author(s) were required to supply signed patient consent prior to publication. Author(s) have confirmed that the published article is unique and not under consideration nor published by any other publication and that they have consent to reproduce any copyrighted material. The peer reviewers declared no conflicts of interest.
Footnotes
Acknowledgment
We thank Dr. Michael Atalay, Dr. Stanley Schwartz and Dr. Joe Yammine for help with obtaining and editing photographs.