
Abstract
Hepatosplenic T-cell lymphoma (HSTCL) is a rare and aggressive extranodal T-cell lymphoma that comprises <5% of peripheral T-cell lymphomas. The majority of cases harbor the γδT-cell receptor (TCR), but recently, a few cases have been shown to express the αß TCR. Comparison of these two subtypes (αβ and γδ) shows similar clinicopathologic and cytogenetic features; however, due to the paucity of reported cases, it is not clear whether they are prognostically distinct entities. We report a case of αβ HSTCL with a rather unusual presentation of Coombs'-negative hemolytic anemia. Diagnosis proved challenging due to an unusual blastoid morphology with the absence of typical intrasinusoidal distribution of tumor cells in the bone marrow. This unique case adds to the growing list of this rare subtype of T-cell lymphomas, which warrant urgent attention due to the lack of effective treatment options and dismal prognosis.
Keywords
Introduction
Hepatosplenic T-cell lymphoma (HSTCL) is a rare and aggressive extranodal T-cell lymphoma that comprises <5% of peripheral T-cell lymphomas. 1 It is characterized by extranodal infiltration of malignant mature post-thymic T-lymphocytes in the sinusoids of the liver, spleen, and bone marrow. 1 The majority of cases harbor the βδ T-cell receptor (TCR), but recently, a few cases have been shown to express the αβ TCR. Since the first report of an αβ hepatosplenic T-cell lymphoma (αβ HSTCL) in 2000, 2 <30 cases have been reported in the literature. As the majority of early cases were of the γδ subtype, the entity was initially named “hepatosplenic gammadelta T-cell lymphoma” in the Revised European American Lymphoma (REAL) classification. 3 With increasing reports of new cases with the αβ subtype, the term “HSTCL” is preferred in the current World Health Organization (WHO) Classification. 4 Comparison of the two subtypes 5 shows similar clinicopathologic and cytogenetic features; however, due to the paucity of reported cases, it is not clear whether they are prognostically distinct entities.
We report a case of αβ HSTCL presenting with Coombs'-negative hemolytic anemia. Diagnosis proved challenging due to an unusual blastoid morphology with the absence of typical intrasinusoidal distribution of tumor cells in the bone marrow. This unique case adds to the growing list of this rare subtype of T-cell lymphomas, which warrant urgent attention due to the lack of effective treatment options and dismal prognosis.
Case Report
A male in his 20s from Nepal with no chronic illness presented at our hospital with a one-month history of high-grade fever, skin rash, weight loss, symptoms of anemia, and abdominal pain. He was in fair general condition with normal vital signs except for fever (39 °C). Physical examination revealed a purpuric rash over his trunk and extremities, hepatomegaly (4 cm), and splenomegaly (10 cm) below the costal margin. No lymph nodes were palpable. Initial investigations showed bicytopenia with a platelet count of 36 × 10 3 /μL and hemoglobin 7.0 g/dL with normal mean corpuscular volume of 90 fL (elevated reticulocytes at 233 × 10 3 /μL; reticulocyte index 4.5%, suggesting adequate marrow response). The white blood cell count was 9.7 × 10 3 /μL. A peripheral blood smear showed a leukoerythroblastic picture with several blast-like cells with fine chromatin and one or more prominent nucleoli (Fig. 1). There were also features of hemolysis, including spherocytosis, significant polychromasia, and nucleated red blood cells. The direct antiglobulin test was negative. Coagulation tests and kidney and liver functions were within normal limits. Serum lactate dehydrogenase was high (1,169 IU/L; normal up to 225) and haptoglobin was low (<3 mg/dL). Blood and urine cultures were repeatedly normal. Serologies and polymerase chain reaction (PCR) tests for hepatitis B and C virus, human immunodeficiency virus, cytomegalovirus, Epstein-Barr virus, Leishmania, and human T-lymphotropic virus type 1 were negative. Echocardiography showed no vegetation. Computed tomography (CT) scan showed marked splenomegaly with a span of 21.8 cm associated with arterial hypoperfusion (perhaps explaining the abdominal pain). Enlarged axillary, porta hepatis, mesenteric, and para-aortic lymph nodes were also noted (the largest lymph node was 19 × 13 mm in the abdomen). A whole-body FDG PET/CT scan showed moderate uptake in the left axillary and upper retroperitoneal lymph nodes, suggesting lymphoma activity with spleen and bone marrow involvement. The patient underwent a bone marrow aspirate and biopsy examination. The aspirate showed hypercellular marrow with erythroid hyperplasia (mixed normoblastic and megaloblastoid), hypoplastic granulopoiesis, and reduced megakaryocytes. There was marrow infiltration with 18% abnormal pleomorphic cells with patchy distribution, mostly showing blast morphology with primitive chromatin, prominent nucleoli, and irregular nuclear contour. Cohesive clusters of the abnormal cells were also noted (Fig. 2).
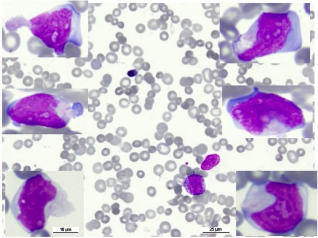
Peripheral blood smear showing a leukoerythroblastic picture with severe thrombocytopenia (Wright stain ×400). Inserts showing examples of the circulating lymphoma cells having blastic morphology; large in size with finely dispersed chromatin and prominent nucleoli with irregular nuclear contour (×1,000).
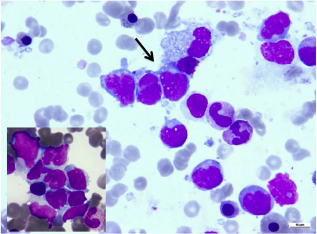
Bone marrow aspirate showing involvement of lymphoma cells with blastic morphology. Insert (bottom left) and arrow show cohesive clusters of lymphoma cells (Wright stain ×1,000).
Flow cytometric analysis (Navios, Beckman Coulter) of the bone marrow aspirate (Fig. 3) revealed a population of abnormal T-cells (18%) of high side scatter expressing CD2, CD3, CD8, and TCR αβ with brighter CD45 and dimmer CD7 than the normal T-lymphocytes with partial loss of CD5. These cells were negative for CD4, CD56, CD16, CD57, CD34, CD1a, and Tdt. Flow cytometry analysis of the TCR V β (Vb) repertoire revealed that the abnormal cells failed to express any of the TCR Vb regions, suggesting monoclonality.
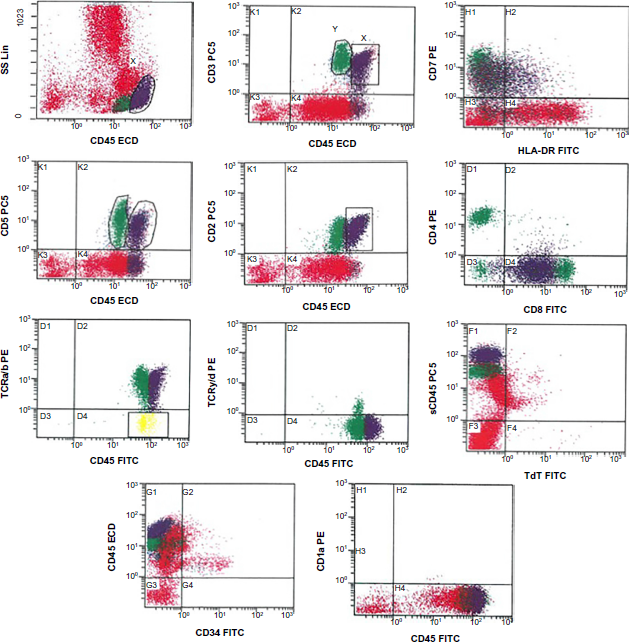
Flow cytometry analysis of bone marrow aspirate at presentation revealed ~18% abnormal T-cells (X gate, purple population) showing immunophenotypic aberrancy (brighter CD45, slight downregulation of CD3 and CD7 with partial loss of CD5 expression) compared with the normal T-cells (gate Y, green population) and expressing CD2, CD8, and TCR αβ. The abnormal T-cells are negative for precursor markers CD34, Tdt, and CD1a.
Immunostains were performed with Autostainer Link 48 and Dako reagents. The immunostains confirmed that the remarkably increased T-cells were positive for CD2, CD3, CD5, and CD8 with loss of CD7 expression (Fig. 4) and negative for CD34, TDT, CD99, Granzyme B, and ALK1.

Bone marrow biopsy showing interstitial infiltration by medium-to-large lymphoma cells, many with irregular nuclear contour (
Cytogenetic analysis of the bone marrow sample revealed normal karyotype, and TCR gene rearrangement performed using peripheral blood showed no evidence of clonality Based on the peripheral blood and bone marrow findings, a diagnosis of αβ mature T-cell lymphoma was made.
After further multidisciplinary discussion, a diagnosis of HSTCL stage IV (αβ subtype) was confirmed. The hyper-CVAD protocol was initiated after discussion at a multidisciplinary conference, and the patient subsequently received the first cycle of treatment, which was complicated by febrile neutropenia and hypotension. He was managed with intravenous antibiotics but continued to have lymphoma-related fevers with persistent massive splenomegaly and no changes in his complete blood count findings. At this point, he also developed a severe headache. Magnetic resonance imaging of the brain showed a small lesion (7 × 9 × 9 mm) in the right temporal lobe, and the possibility of lymphomatous deposition was considered as a likely etiology. A lumbar puncture was not pursued because of refractory thrombocytopenia (<50,000/μL).
Repeated bone marrow examinations after the first cycle of treatment showed increasing abnormal T-cells (53%) with blast morphology scattered in many cohesive clusters. Flow cytometry findings were comparable to those at presentation except for loss of CD5 expression. A second bone marrow aspirate revealed abnormal karyotype with 47,X,-Y, i(7) (q10),+8,+8,add(13)(p11.1) [7]/46,X,-Y,i(7)(q10),+8,add(13) (p11.1) [2]/46,XY[61]. Interval fluorescence in situ hybridization analysis revealed additional signals at 7q31 (D7S486 probe) on chromosome 7 in 15% of the analyzed cells. At this time, PCR showed a clonal TCR gene rearrangement. This along with the other findings confirmed the diagnosis of the rare αβ HSTCL. After further discussion, a second cycle of hyper-CVAD was initiated, which was complicated by Enterobacter cloacae sepsis that was treated with meropenem.
After two cycles of hyper-CVAD therapy, bone marrow evaluation showed 17% neoplastic T-cells (similar to 18% at initial diagnosis) and flow cytometry showed the same immunophenotype (as at initial diagnosis) except for persistent loss of CD5. Interval PET-CT imaging showed no significant change compared to baseline, suggesting the absence of response to hyper-CVAD therapy. Given the poor prognosis of the disease, the patient was offered salvage therapy with regimens containing high-dose cytarabine and platinum analogs (DHAP or ESHA), followed by autologous stem cell transplantation. The poor prognosis of the disease and its refractoriness to chemotherapy along with the salvage therapy options were discussed with the patient. He decided to return to his home country and was subsequently discharged.
Discussion
This case of αβ HSTCL confirms several of the previous findings of this rare disease5,6 and also illustrates some atypical features that make it a diagnostic challenge, especially given its extremely low incidence.
The peak incidence of HSTCL is in adolescents and young adults with a median age at presentation of 20 years and with a gender ratio of 9:1 (male:female). The largest case series of αβ HSTCL (14 cases) was reported by Macon et al. 5 who showed no significant clinicopathologic and cytogenetic difference between the αβ and γδ subtypes, except for female preponderance, older median age, and variable age distribution in the former. Up to 20% of γδ HSTCL cases arise in the background of chronic immune suppression, particularly solid organ transplantation,7-9 a finding denied by Nagai et al for the αβ subtype. 6 It is worth mentioning that our patient had no significant past medical history. In this context, more cases are needed to fully characterize this disease as a separate entity.
HSTCL usually presents with marked splenomegaly and hepatomegaly, but without lymphadenopathy 1 Interestingly, our patient had no palpable lymphadenopathy, but CT and PET imaging unveiled axillary, porta hepatis, and retroperitoneal lymphadenopathy. Patients also commonly have B symptoms, such as fever, fatigue, and weight loss. 1 Abnormalities in liver function tests may be seen in half of the patients. 1
Thrombocytopenia is also a common feature of this disease with associated leukopenia and anemia. Autoimmune hemolytic anemia (“warm” type and cold agglutinin disease)10,11 and immune thrombocytopenic purpura 12 have been reported as initial manifestations of the disease in rare cases. Cytopenias may result from hypersplenism, bone marrow failure, cytokine-mediated hemophagocytic histiocytosis, 13 or a combination of these mechanisms. The pathogenesis of autoimmune hemolytic anemia in HSTCL is unknown, although neoplastic T-cells as mediators of autoimmune disease have been proposed as a potential explanation. 14 Our patient initially presented with a picture of Coombs'-negative hemolytic anemia. It is also worth noting that 5%-10% of autoimmune hemolytic anemias are Coombs’ negative, 15 and several causes for this phenomenon have been identified, which include hemolysis by natural killer (NK) cells independent of antibody, small number of subthreshold immunoglobulin (Ig) G attached to red blood cells, low-affinity autoantibodies, IgA autoantibodies, and IgM autoantibodies. 14 Coombs'-negative hemolytic anemia was previously reported in one case of γδ HSTCL. 16 To the best of our knowledge, this is the first reported case of αβ HSTCL with such a presentation.
Morphologically, HSTCL is characterized by a monotonous neoplastic infiltrate consisting of medium-sized lymphocytes with a moderate cytoplasm and irregular cellular borders. The nucleus is typically oval with less condensed chromatin than small lymphocytes. The splenic red pulp is expanded with infiltration of cords and sinuses associated with atrophy of the white pulp. Liver biopsy also shows an intrasinusoidal infiltrative pattern. 1 The bone marrow is frequently involved in the disease. 1 Trephine biopsy typically shows a hypercellular marrow with characteristic selective infiltration and expansion of the sinuses, which is a useful diagnostic criterion.17,18 However, with progression, the pattern of bone marrow involvement becomes increasingly interstitial, 16 which was likely the case in our patient, thereby posing a diagnostic challenge. Furthermore, morphology of the tumor cells was predominantly blastoid, another atypical feature of HSTCL that is typically associated with disease progression.16,17,19 Cohesive clustering of lymphoma cells was another unusual finding. These atypical findings, along with the possibility of false-negative initial karyotype and TCR rearrangement studies, delayed the definitive diagnosis. The false-negative result is possibly related to sample dilution, leading to loss of signal below the detection limit. Given the blastoid morphology of the cells, acute lymphoblastic leukemia/lymphoma was considered in the differential diagnosis. However, the negativity for precursor markers (CD34, TdT, and CD99) excluded this possibility. Other differential diagnoses considered included splenic marginal zone lymphoma, T-cell large granular lymphocytic leukemia, and aggressive NK/T-cell leukemia, which were excluded because of the inappropriate morphology and immunophenotype.
HSTCL typically has the following phenotype: CD2+, CD3+, CD4–, CD5–, CD8–, CD16–, CD56+/-, and TCRaβ/γδ+.4,20 Frequently, loss of CD3, CD5, and/or CD7 (CD5 lost in this case) occurs with disease progression. The lymphoid infiltrate in our case was CD8+, which is reported in a minority of cases.4,20 Interestingly, Macon et al. 5 showed that the αβ type was more likely to be CD8+ when compared to the γδ type. NK cell markers may also be usually expressed with an inactive cytotoxic profile (TIA+, Granzyme-, and Perforin-); however, our case did not show any.
Most γδ HSTCLs are associated with the presence of an isochromosome 7q (i[7][q10]) with associated abnormalities such as trisomy 8 and loss of chromosome Y. The isochromosome 7q has also been demonstrated in αβ HSTCL cases.5,11,21 All the abovementioned karyotypic abnormalities were observed in our patient. The biological significance of the isochromosome 7q has not been worked out to date and is not entirely specific to this disease.
The prognosis of HSTCL, αβ or γδ type, is extremely poor with a median survival of less than one year. 5 According to the International Peripheral T-Cell and Natural Killer/T-Cell Lymphoma study, 22 the five-year overall survival was only 7% and five-year failure-free survival was 0%, this being the worst among all subtypes of T-cell lymphomas. Management strategies are quite varied with no consensus first-line therapy regimen. The majority of patients respond to CHOP or CHOP-like regimens but quickly relapse with very few long-term survivors. 19 Therapies that are quite effective in curing a substantial number of patients with other aggressive lymphoid malignancies such as diffuse large B-cell lymphoma have been shown to be ineffective for HSTCL. Several case reports have studied the efficacy of new treatments, such as bortezomib, alemtuzumab, and allogeneic bone marrow transplantation,23–26 but more efforts are still needed to treat this rare but devastating disease.
Author Contributions
Conceived and designed the experiments: FI. Analyzed the data: FI, AA, DS. Wrote the first draft of the manuscript: VS. Contributed to the writing of the manuscript: FI. Agreed with manuscript results and conclusions: AS. Jointly developed the structure and arguments for the paper: FI, VS. Made critical revisions and approved the final version: HO, RT. All authors reviewed and approved the final manuscript.
Footnotes
Acknowledgments
The authors gratefully acknowledge the contributions of all the staff of the flow cytometry and cytogenetics labarotories at the National Center for Cancer Care and Research in specimen processing and data collection.