
Abstract
Cases of extraskeletal Ewing sarcoma (EES) originating primarily within the spinal epidural space, are very rare and have a very poor prognosis. There is no standard therapy for this disease.
We report the case of a 23-year-old man presenting with symptoms of back pain and numbness of both legs for 10 days. Imaging studies revealed a dorsal soft-tissue, extradural mass at the T8–9 vertebral level. The patient underwent a laminectomy and complete excision of the tumor. The EES diagnosis was confirmed by histologic analysis including immunohistochemistry and by presence of the EWS-ERG due to the t (21: 22) (q22: q12) chromosomal translocation by a reverse transcriptase-polymerase chain reaction (RT-PCR). This is the first report of spinal epidural EES with presence of the EWS-ERG fusion transcript. Post-operatively, the patient received aggressive adjuvant chemotherapy and radiotherapy.
At 63 months after surgery, the patient is without clinical or radiological evidence of recurrent or metastatic disease.
Early discovery of EES and a complete resection followed by the aggressive treatment with radiation and chemotherapy may improve disease-free and overall survival.
Introduction
Ewing sarcoma (ES) and peripheral primitive neuroectodermal tumor (pPNET) are closely related malignant, small, round cell tumors, and usually found in long bones. They rarely have an extraosseous origin. Based upon reports in the literature, it is exceptionally rare to find them originating primarily within the spinal epidural space; only 34 such cases have been reported to date [1, 5, 7–10]. Most patients are between 10 and 20 years old and male. A diagnostic delay is apt to occur because it often causes non-specific symptoms, and therefore its prognosis is often not as good as would be expected for ES [5, 7, 10]. Primary spinal epidural EES should be suspected whenever young patients present with back pain and/or radicular pain, have abnormal neurological findings and an extradural mass is detected on MRI. A complete resection at an early stage and the use of aggressive radiotherapy and chemotherapy are indispensable to achieve local control and prevent distant metastasis [5].
The differential diagnosis includes other small, blue round cells tumors [1]. In ES/pPNET, the detection of the chromosomal translocation by RT-PCR is useful for making an accurate diagnosis, in addition to the usual histochemical stains and immunohistochemistry.
Case Report
A 23-year-old man complained of back pain which had lasted for 10 days followed by the onset of numbness in the anterior and posterior aspect of both legs for 2 days prior to presentation at our institution. His motor and reflex findings were normal and there was no bowel or bladder dysfunction. Haematologic tests were in the normal range. His past and family history were unremarkable.
Magnetic resonance imaging (MRI) was performed on the day he presented to our hospital, and it revealed an epidural mass extending from the T8–9 vertebrae and compressing the cord dorsally (Fig. 1A). No other vertebral abnormality was seen in the cervical and lumbar region. He was admitted, and the next day, to confirm whether the lesion arose from bone or not, and the relationship between it and the dura mater, myelography and computed tomography (CT) scanning were performed. A postmyelogram CT scan revealed an epidural mass at the level of T8–9, which displaced the spinal cord ventrally. No osseous erosion or destruction was evident (Fig. 1B).

(
Neither a primary tumor nor a metastasis was detected elsewhere in the body.
Because the numbness and weakness of both legs progressed over the two days following his hospitalization, an urgent laminectomy of T7–10 and a complete resection of the tumor mass was performed. Intraoperativery, no obvious adhesion to the dura mater or infiltration into the surrounding tissues was seen. In the resected specimen, the tumor was reddish-grey in color, and no obvious bony involvement of the ventral aspect of the lamina was seen grossly (Fig. 2).
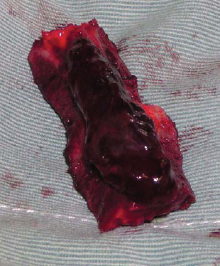
In the resected specimen, the tumor was reddish-gray without adhension to the dura mater. No obvious cortical destruction was seen grossly.
Histopathologically, the tumor was composed of a uniform sheet of small cells, round to oval in shape with irregularly shaped chromatic nuclei surrounded by scanty cytoplasm. No special cellular arrangement, such as rosettes was seen, nor was there any evidence of differentiation (Fig. 3). Periodic acid-Schiff (PAS) staining showed abundant intracytoplasmic glycogen. Immunohistochemical staining was positive for CD99 (MIC 2) and vimentin. Furthermore, RT-PCR using the primers described by Jin [6] was performed, and it confirmed the presence of the EWS to ERG fusion transcript indicative of the t (21; 22) (q22; q12) translocation characteristic of ES/pPNET (data not shown). The diagnosis was thus extraskeletal Ewing sarcoma.

Photomicrograph showing diffuse proliferation of sheets of cells with irregularly shaped chromatic nuclei with scanty pale cytoplasm. (hematoxylin and eosin, ×160).
Beginning two weeks following surgical intervention, the patient received six courses of combination chemotherapy with VAIA (vincristine, doxorubicin, ifosfamide, and actinomycin-D) followed by four courses of ICE (ifosfamide, carboplatin and etoposide) combined with external beam radiotherapy to a dose of 44Gy in 22 fractions over 4 weeks using the same schedule according to a decision of our treating center. The patient completed the radiotherapy and chemotherapy, and 63 months after diagnosis, he is without clinical or radiological evidence of recurrent or metastatic disease and no neurologic deficit.
Discussion
ES/pPNET is a relatively common small round cell sarcoma of bone that occurs predominantly in the metaphysis of long bones in skeletally immature patients. It accounts for 6% to 8% of all malignant primary bone tumors [4]. EES is rare, and the exact incidence of EES is not known but it varies between 10% and 13% osseous ES [3]. The most frequent sites of occurrence of EES are the chest wall, lower extremities, trunk, and pelvis [12]. However, these tumors rarely originate within the epidural space of the spine. In the English literature, 34 additional cases of EES/pPNET arising primarily from within the spinal canal have been reported [1, 5, 7–10]. In a review of 34 cases, the mean age at diagnosis of the patients with spinal epidural EES was 19.2 years and 18 of 34 cases were in their second decade of life at diagnosis; most (69.7%) patients were male.
The mean diagnostic delay was 4.9 months because of the non specific symptoms at onset. The most common symptoms included back pain and/or radicular pain in all patients, paresis of the leg(s) in 69.7% of patients, sensory disturbances, and bladder and bowel dysfunction caused by compression. These might be mistaken for various orthopaedic conditions that might also produce similar symptoms [10]. Therefore, MRI examination is indispensable, and the findings should be always distinguished from intervertebral disk herniation, and other benign or malignant tumors [9]. From its histological appearance, lymphoma and rhabdomyosarcoma should be excluded and the possibility of melanoma and neuroblastoma should be considered in the differential diagnosis. Immunohistochemical staining for the MIC-2 gene product is helpful to differentiate ES/pPNET from other malignancies. Moreover, genetic analysis, including fluorescent in situ hybridization (FISH) or RT-PCR, is very helpful when morphology is not conclusive or when the tumors are present in unusual clinical settings [2, 11]. In our case, in addition to immunohistochemical analysis, the EWS-ERG fusion was detected by RT-PCR, so that the diagnosis of ES was confirmed. In more than 95% of ES/pPNET cases, the gene fusion is EWS-FLI1 (90% to 95%), due to the t (11; 22) (q24; q12), or EWS-ERG (5% to 10%) due to the t (21; 22) (q22; q12) [13]. This is the first report of spinal epidural EES with presence of the EWS-ERG fusion transcript.
Generally, ES/pPNET are exquisitely radiosensitive and chemosensitive, and radiotherapy and chemotherapy are often employed pre- and post-operatively. In spinal epidural EES/pPNET, it is believed that these adjuvant therapies also are indispensable after a laminectomy with tumor resection to avoid a local recurrence or distant metastasis [10]. In the literature, the recommended dose of radiation therapy used in patients with spinal epidural EES/pPNET varies between 30 Gy and 56 Gy, and it is difficult to define the optimum radiation dose because the survival rate does not always depend on it [5]. In our case, 44Gy of radiation therapy after surgery was administered.
The chemotherapeutic agents most commonly used are vincristine, doxorubicin, cyclophosphamide, ifosfamide, and actinomycin-D. In our case, we used combination chemotherapy with six courses of the VAIA regimen as the induction phase and four courses of the ICE regimen as the maintenance phase. Some investigators have reported the usefulness of high-dose chemotherapy with peripheral blood stem cell rescue [5, 9], and the VAC (vincristine, doxorubicin, cyclophosphamide) regimen alternating with the ICE regimen for six cycles [10] for this condition.
The clinical results of spinal epidural EES/pPNET are very poor, even if adjuvant therapies are administered. The 5-year survival rate for spinal epidural EES/pPNET has been reported to be between 0% and 37.5% [7]. This result is low in comparison with the 5-year survival of EES which is between 38% and 67% [10]. This poor prognosis may result from the high rate of incomplete resections [5]. Of the 34 reported cases, 14 patients (41.2%) died of their disease at a median follow-up period 21.3 months and in 17 patients (50%), the tumors were only partially resected, due to infiltration or adhesion of the surrounding tissues. In our case, fortunately, no evidence of infiltration or adhesion of the surrounding tissues was seen, so that a complete resection could be performed.
In conclusion, although EES/pPNET arising primarily from within the spinal epidural space is very rare, it should be included in the differential diagnosis of young patients presenting with a history of back pain and radicular pain. The prognosis of this disease is very poor, due to the high rate of incomplete resection. In our case, a complete resection would be performed because there was early recognition of the lesion on MRI. RT-PCR detected the presence of the EWS-ERG fusion transcript, which helped to confirm the diagnosis.