
Abstract
Hypertrophic cardiomyopathy (HCM) is a global disease with cases reported in all continents, affecting people of both genders and of various racial and ethnic origins. Widely accepted as a monogenic disease caused by a mutation in 1 of 13 or more sarcomeric genes, HCM can present catastrophically with sudden cardiac death (SCD) or ventricular arrhythmias or insidiously with symptoms of heart failure. Given the velocity of progress in both the fields of heart failure and HCM, we present a review of the approach to patients with HCM, with particular attention to those with HCM and the clinical syndrome of heart failure.
Introduction
In 1957, Donald Teare of St. George's Hospital in London described eight cases of asymmetric septal hypertrophy seen on autopsy with some bewilderment, noting that the tumors had “occurred in a group where cardiac incapacity … is rare.” He ultimately described “the eight tumours under discussion as hamartomata.” 1 In 1959, Morrow and Braunwald published a case series of three patients with a clinical syndrome mimicking aortic stenosis. In the first two patients, “significant pressure gradients were demonstrated preoperatively but … no anatomic site of outflow obstruction could be detected at the time of open-heart operation.” When the third patient was taken to surgery, a discrete hypertrophy of the subaortic interventricular septum was resected, resulting in resolution of the stenosis and improvement of the patient's symptoms. 2 By the time of their 1964 case series of 64 patients with a condition they termed idiopathic hypertrophic subaortic stenosis,3,4 Braunwald's group had launched a campaign now spanning more than five decades of clinical care and research of the condition we now know as hypertrophic cardiomyopathy (HCM).
It is now recognized that HCM is a global disease with cases reported in all continents, affecting people of both genders and of various racial and ethnic origins. 5 Despite varied presentations, the disease has similar genotypic abnormalities and the unifying phenotypic expression of left ventricular hypertrophy (LVH). In a variety of geographic locales, the incidence of HCM is similar, approximately 1 in 500 (0.2%) of the general population. 6 In the USA alone, this incidence estimates the total number of affected people at approximately 600,000. This relatively high prevalence stands in contrast to the comparatively low rate of recognition and frequent delay in diagnosis of HCM, particularly in women 7 and people of African-American origin. 8
Genes responsible for HCM, and their symbols, loci, and estimated frequency.

The clinical sequelae of HCM include atrial and ventricular arrhythmias; sudden cardiac death (SCD); left ventricular (LV) outflow tract obstruction, which is often dynamic and variable and may lead to syncope; and heart failure in the setting of either preserved (HFpEF) or reduced (HFrEF) systolic function.11,12 There exist excellent and comprehensive reviews of HCM and its management on a broad scale.13,14 However, given the velocity of progress in both the fields of heart failure and HCM, we feel a focused review on the approach to patients with HCM and the clinical syndrome of heart failure is warranted. In this article, we aim to provide a review of the clinical course, diagnosis, and therapies for patients with HCM, with particular focus on the heart failure syndrome.
Clinical Presentation and Diagnosis
Natural History
In the nascent years of the investigation into HCM, cohort studies of patients with the disease painted a dire picture of prognosis – with mortality estimated from 5 to 6% annually. 15 This view of HCM as a singularly unfavorable disease, though, was based on patients referred only to tertiary care centers; older patients and those with clinically stable disease were systematically underrepresented.11,16
Based on more recent, balanced overviews of patients with HCM, the annual mortality for patients with HCM is estimated at 1% per year.11–14 It appears that both extremes of the epidemiologic scale, children <10 years and adults >80 years, are increasingly represented in cohort studies as our ability to diagnose HCM with non-invasive imaging has become more refined and our therapeutic approaches provide more opportunities for advanced longevity.11–13,17
Though the prognosis of HCM appears better than previously believed, many patients with HCM can suffer from a variety of symptoms. First, a subset of patients experiences sudden death in the absence of antecedent symptoms. Second, 20% of patients develop atrial fibrillation (AF),11,12 which exacerbates other accompanying clinical symptoms and carries a risk of embolic stroke. Third, the patients can experience anginal chest pain because of microvascular ischemia from a (blood) supply and demand (excess myocardium) mismatch or, rarely, myocardial bridging. 18
Finally, and the focus of this review, the patients may suffer from progressive heart failure. This includes patients with preserved or reduced ejection fraction as well as those with or without outflow tract obstruction. Any patient with HCM may eventually progress to end-stage heart failure with reduced LV systolic function. 14 The evolution to this dilated hypokinetic phenotype of HCM is believed to occur progressively as myocardial fibrosis and other adverse remodeling changes accumulate.19,20 It is possible that this evolution is predisposed by specific genetic mutations. 21
The evolution of severe heart failure (New York Heart Association [NYHA] functional class III or class IV) occurs in 10-20% of patients with HCM. 22 While these symptoms can occur at any age, they are most frequently seen in middle-aged adults. Women tend to have more severe symptoms of heart failure occurring later in life. 7 The risk of heart failure is augmented by the presence and degree of outflow tract obstruction. 23 Heart failure can occur in one-third of patients who have HCM without outflow obstruction, though it is less common. 24 Further risk factors of heart failure include the presence of AF 25 and diastolic dysfunction, though non-invasive measures do not reliably predict LV filling pressures in HCM. 26 Notably, LV wall thickness is not predictive of progressive symptoms of heart failure. 27
Physical Examination
It is increasingly recognized that many patients with HCM have a normal physical examination. Below, we summarize the classical examination findings with the caveats that (a) many of these findings depend on the presence of an outflow obstruction, and (b) because of the dynamic nature of outflow obstruction in HCM, even patients with the presence of an inducible outflow gradient may have normal findings at the time of the examination.
Inspection of the precordium may reveal a prominent parasternal lift (representing right ventricular enlargement because of left heart disease) or apical impulse. The internal jugular veins may have a prominent a wave (Fig. 1) and are often elevated in patients with heart failure. Palpation of the point of maximal impulse may reveal a sustained and enlarged LV apical impulse, a presystolic apical impulse representing atrial systole in a non-compliant ventricle, or in rare cases, a systolic thrill at the apex or lower left sternal border. Palpation of the carotid pulse may expose a bifid, brisk waveform in patients with significant outflow obstruction representing the initial rapid phase of ejection followed by a second decelerated phase caused by the mid-systolic obstruction and partial aortic valve closure (spike and dome; Fig. 1). Paradoxical splitting of the second heart sound on auscultation may occur in patients with significant LV obstruction because of the delayed closure of the aortic valve.

Venous and arterial waveforms in HCM. JVP waveform in HCM showing an augmented a wave. Carotid impulse tracing in HCM demonstrating the spike (red arrow) and dome (blue arrow) pattern. Adapted from Goldstein JA. Cardiac tamponade, constrictive pericarditis, and restrictive cardiomyopathy. Curr Probl Cardiol. 2004;29(9):503-567.
Two murmurs are often cited as being present in patients with HCM. The first murmur is because of systolic anterior motion (SAM) of the mitral valve leading to poor leaflet coaptation and mitral regurgitation (Fig. 2B and C). This causes a mid-systolic murmur at the apex radiating to the axilla (though this may be variable because of an eccentric direction of the regurgitant jet). The second murmur is because of turbulent flow through the outflow tract and is present as a mid-systolic, crescendo-decrescendo murmur, often loudest at the left lower sternal border, which can mimic the murmur of aortic stenosis. Maneuvers may enable the differentiation between the two entities. As opposed to aortic stenosis, maneuvers that reduce preload (eg, Valsalva, squat-to-stand, dehydration) will cause an augmentation of the murmur intensity in patients with HCM. Alternatively, maneuvers that increase preload (stand-to-squat or passive leg raise) will lead to a reduction in murmur intensity in HCM. An S4 gallop may be present in patients with HCM in sinus rhythm because of atrial systole against a poorly compliant LV, and in patients with HFrEF, an S3 may be heard as well.
Imaging of HCM. (
Electrocardiogram (ECG)
Electrocardiographic abnormalities (Fig. 3) are nearly ubiquitous in HCM patients, occurring in all but 5% of patients in one published cohort.
28
While its high sensitivity makes the ECG an optimal screening test, the abnormalities are varied and non-specific. Typically, the ECG reveals prominent voltages with localized or widespread repolarization abnormalities. Other abnormalities include prominent inferior or lateral Q-waves, left axis deviation, and p-wave abnormalities including left or right atrial abnormalities. Pseudo-delta waves may also be seen, mimicking the preexcitation syndromes (eg, Wolff–Parkinson–White syndrome).
29
ECG of a 51-year old patient with HCM. Note the prominent precordial voltage, widespread repolarization abnormalities, Q-wave in the lateral lead (aVL), and p-wave abnormality suggesting left atrial enlargement.
Laboratory Studies
In patients with HFrEF, B-type natriuretic peptide (BNP) levels have been championed as both diagnostic of a congested state and prognostic of HF mortality. 30 In HCM patients, one study found a trend toward higher levels of BNP in patients with symptomatic heart failure that increased with heart failure severity. 31 However, the study did not control the degree of LV wall thickness, which was independently associated with BNP levels regardless of heart failure symptoms or severity, and there was considerable overlap of BNP levels in patients with mild, moderate, and severe heart failure. 31 Thus, relying on BNP levels alone to diagnose heart failure in patients with HCM may be of limited clinical utility.
Serum troponin I and T assays are prognostic in HCM. One study established that higher levels of high-sensitivity troponin T were correlated with increased LV wall thickness and worsened diastolic dysfunction. 32 This biomarker has also been associated with an elevated risk of cardiovascular deaths, heart failure admissions, sustained ventricular tachycardia, embolic events, and progression to NYHA functional class III or class IV status over a four-year follow-up period. 33
Imaging: echocardiography and cardiac magnetic resonance imaging (MRI)
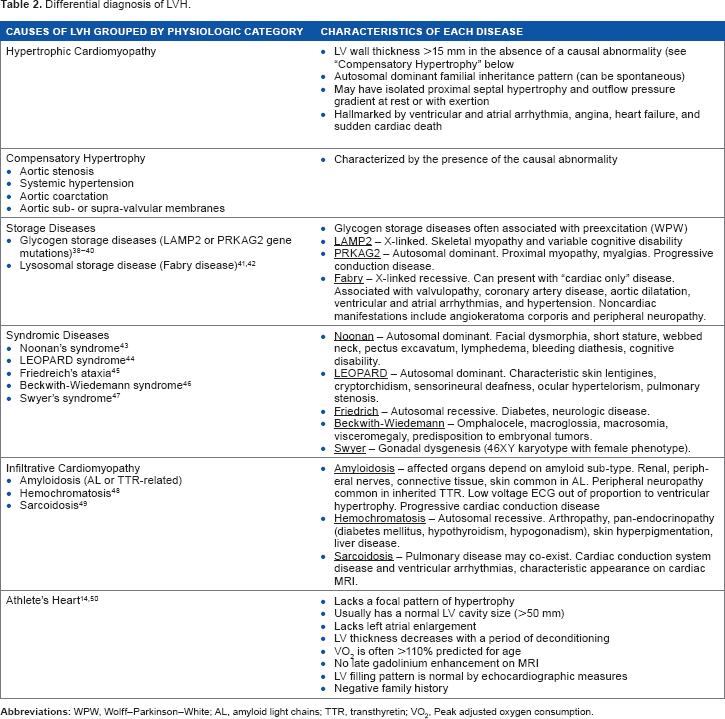
Imaging takes a central role in establishing both diagnosis and prognosis in HCM. While family history, symptoms, physical examination, and ECG can all be suggestive factors, none are necessary or sufficient to diagnose a patient with HCM. The presence of LVH (>15 mm LV wall thickness in diastole) in the absence of another cause confirms the diagnosis11–14 (Fig. 4). The two most common imaging modalities employed are two-dimensional (2D) echocardiography and tomographic high-resolution cardiovascular MRI (Fig. 2).
Schematic representations of a normal heart (left) and a heart with HCM (right). Reproduced with permission from Nishimura R. Hypertrophic cardiomyopathy: a patient perspective. Circulation. 2003;108:e133-e135.
One advantage of 2D echocardiography is the ability to evaluate the ventricular, valvular, and outflow states under various provocative maneuvers, most commonly exercise. Using continuous wave, pulse wave, and color Doppler techniques, echocardiography provides accurate estimates of outflow tract gradients and valvular regurgitation. Outflow tract gradients of 30 mmHg or more at rest are independent predictors of heart failure symptoms. 23 In the absence of a resting gradient, the discovery of an elevated gradient with exercise may explain exertional symptoms and guide therapeutic decisions. Historically, pharmacologic agents (dobutamine, amyl nitrate, isoproterenol) or the Valsalva maneuvers 4 were employed to provoke outflow tract obstruction. However, they have progressively lost favor because of concern that they may not reflect the physiologic state during the patient's real-world activities. 24 Exercise remains the preferred provocative method whenever possible.
Tomographic high-resolution MRI (Fig. 2D) is often superior to 2D echocardiography in evaluating LVH in atypical locations, eg, the anterolateral free wall, apex, or posterior septum. 34 Similarly, MRI appears superior in identifying patients with apical aneurysms, which carry increased risk of ventricular arrhythmias and possibly thrombosis. 35 MRI can also quantify the degree of myocardial fibrosis with late gadolinium enhancement (LGE). A high burden of LGE in patients with HCM (≥20% of LV myocardium) may be a predictor of SCD. 36 One study also suggested that the presence of substantial LGE may identify patients progressing toward end-stage disease, particularly in the presence of a low-normal Left Ventricular Ejection Fraction (LVEF). 37
Endomyocardial Biopsy
The routine use of endomyocardial biopsy for patients with a typical HCM phenotype is controversial. Historically, endomyocardial biopsy in patients with suspected HCM has not been recommended because of the presumed low incidence of this storage or infiltrative disease.
Differential diagnosis of LVH.
Screening
HCM is an autosomal dominant disorder, and the first-degree relatives of an affected individual should undergo screening with a detailed history, physical examination, ECG, and echocardiography.11–14 The AHA recommends screening starting at age 12 or earlier if the child has a growth spurt or early signs of puberty, is symptomatic, is involved in high-intensity sports, or has a high-risk family history of SCD. 11 The European Society of Cardiology recommends screening beginning at age 10. 12 From age 12 to 18 years, the first-degree relatives should undergo yearly screening with ECG and echocardiography. After 18 years of age, screening can be spaced to every five years for the asymptomatic individual.11,12 Genetic testing of family members is generally reserved for cases where the genetic mutation is known in the index case.11,12 Patients and their families should receive genetic counseling as part of their evaluation. 11
Therapy
Lifestyle Changes
In patients with HCM, attention to specific lifestyle changes can help mitigate symptoms and may reduce the risk of SCD. These patients should avoid volume depletion, since this will worsen the outflow gradient and may lead to syncope or SCD because of transient systemic hypoperfusion. Similarly, even moderately intense physical activity may lead to syncope or SCD. During exercise, not only is cardiac inotropy and chronotropy augmented, but systemic vascular resistance (SVR) decreases without the ability to augment cardiac output because of outflow obstruction resulting in systemic hypoperfusion. It is recommended that patients with HCM should not participate in most competitive sports with the possible exception of those that involve low intensity.11,12,53
Pharmacologic Therapy
Despite having the benefit of nearly 60 years of clinical and research experience with HCM, a few high-quality, adequately powered trials of pharmacologic therapies exist. 54
Beta-blockers
Beta adrenergic blockers remain an evidence-based mainstay of therapy for HCM, both with and without outflow obstruction at rest. They are one of the most studied pharmacologic therapies for HCM and have been shown to reduce physiologic outflow obstruction, angina, dyspnea on exertion, and the risk of ventricular arrhythmias. These effects are mediated by the reduction in heart rate leading to increased diastolic filling time, decreased inotropy, and possibly a reduction in ventricular stiffness induced by the sympatholytic effects of beta-blockade. 54
Five different beta-blockers have been used in 12 studies in HCM. 54 Propranolol was first studied in four early trials.55–58 One trial compared nadolol (beta-blocker) to verapamil (non-dihydropyridine calcium channel blocker) and found nadolol to be superior in terms of symptomatic relief. 59 Other studies have shown that in patients with an inducible outflow obstruction, bisoprolol is effective in reducing or abolishing any gradient on provocation. 60 While head-to-head studies of beta-blockers are lacking, it is accepted that non-vasodilating beta-blockers should be favored in HCM patients with obstruction to avoid exacerbating the outflow gradient. 12
It remains undefined if prolonged beta-blocker therapy impacts the natural history of patients with HCM, 54 but they remain the current standard of care in symptomatic patients both with and without outflow obstruction.
Non-dihydropyridine calcium channel blockers
The non-dihydropyridine calcium channel blockers (verapamil and diltiazem) are used similar to beta-blockers to reduce cardiac chronotropy and inotropy, leading to improved diastolic filling, reduced outflow gradient, and improved perfusion of the subendocardium. Verapamil remains the single most studied drug in HCM patients. 54 While studies have shown improvement of surrogate clinical outcomes such as LV diastolic parameters on echocardiogram, myocardial ischemia on photon emission exercise testing, and reduced outflow gradients,16,61,62 there is currently no evidence that verapamil or diltiazem improves quality of life or reduces the risk of SCD or heart failure in HCM patients. Indeed, the current HCM guidelines suggest caution in using the non-dihydropyridine calcium channel blockers in patients with either severe outflow obstruction (because of their vasodilatory effect in the peripheral vasculature) 54 or severe heart failure in non-obstructive disease (because of negative inotropy). With these caveats, these drugs are optional for HCM patients.
Disopyramide
Disopyramide is a Vaughan-Williams class IA antiarrythmic medication and has been shown to reduce outflow gradients and improve symptoms in patients with outflow obstruction.63–65 In fact, it is the only drug to date proven to improve outflow gradients at rest. 66 Its side effect profile, however, is more significant than either the beta-blockers or non-dihydropyridine calcium channel blockers and includes risk of QTc prolongation and anticholinergic side effects. 54 Despite these concerns, with careful monitoring, disopyramide remains a reasonable pharmacologic option for patients who remain symptomatic with high outflow gradients and for patients in whom beta-blockers and (or) calcium channel blockers have failed. There is no known role for disopyramide in HCM patients without an obstructive gradient.
Angiotensin-converting enzyme (ACE)-inhibitors, angiotensin receptor blockers (ARBs), and aldosterone receptor antagonists.
Most experts do not endorse the use of ACE-inhibitors or ARBs for patients with HCM and an elevated outflow gradient at rest. The additional reduction in afterload may serve to augment the outflow gradient and worsen symptoms or may lead to syncope. It is important to closely monitor patients with non-obstructive HCM after the initiation of an ACE-inhibitor or ARB, as these medications may cause a clinically significant gradient.
However, in non-obstructive disease, there is provocative early data that ACE-inhibitors and ARBs may be beneficial for patients with HCM. In transgenic mouse models of HCM, angiotensin II appears to augment myocyte disarray and interstitial fibrosis. 67 It is known that increased fibrosis has important functional and prognostic values. 68 Both findings suggest that inhibition of an up-regulated renin-angiotensin-aldosterone system (RAAS) could be beneficial for patients with HCM.
Clinical data in humans with HCM are limited. One study showed that the combined effects of intracoronary enal-april and sublingual captopril can augment coronary flow reserve. 69 Four small pilot trials studying ARBs (candesartan, losartan, and valsartan) in non-obstructive HCM suggest a benefit for LV function and retarded progression of hypertrophy,70–73 though these results have not been correlated with clinical outcomes. It also appears that valsartan can suppress the synthesis of type I collagen in HCM patients, potentially reducing fibrosis. 70 A further study is required to establish if there is a clinical benefit for symptomatic patients with non-obstructive HCM.
Aldosterone has been implicated as a possible contributor to myocyte hypertrophy, disarray, and fibrosis in HCM. 67 It has been noted that myocardial aldosterone levels are significantly increased in HCM when compared to controls.68,74 Similar to the studies of ARBs, rodent models of HCM have demonstrated a decreased degree of myocardial fibrosis and myocyte disarray when exposed to aldosterone receptor antagonists. 75 Though the use of aldosterone antagonists in patients with HFpEF has been largely disappointing in clinical trials, 76 a further investigation of this class is needed in patients with the distinct clinical entity of HCM.
Antiarrythmic Drugs
There exists no randomized data that antiarrhythmic drugs reduce the risk of ventricular arrhythmias. Sotalol may carry more antiarrhythmic benefits than other drugs of the same class, yet it must be used with extreme caution in patients with more than moderate hypertrophy. In one large cohort, no patients on sotalol experienced SCD over a seven-year follow-up period. 77 Notably, pharmacologic therapy with amiodarone or high-dose beta-blockers has not been proven to change the risk of SCD in HCM patients. 77
Management of AF
AF is the most common arrhythmia in HCM, occurring in 20-30% of patients, and often unmasks or exacerbates symptoms of heart failure.13,14 As in the general population, management of AF is composed of two primary aims: rate versus rhythm control and mitigating the risk of thromboembolism.
Similar to any patient, if an HCM patient with AF is hemodynamically unstable, he/she should undergo emergent direct current cardioversion. 12 In the stable HCM patient with AF, factors such as patient symptomatology and preference should be taken into account when deciding between a rate or rhythm control strategy. There are no prospective randomized studies comparing patient outcomes between each strategy. For ventricular rate control, beta-blockers or non-dihydropyridine calcium channel blockers (together or in combination) should be used. 12 In patients with outflow obstruction, digoxin should be avoided.11,12 For patients in whom rate control is unsuccessful, amiodarone has had moderate success in maintenance of sinus rhythm. 78 Flecainide, propafenone, and the IC antiarrhythmic medications are contraindicated because of their proarrhythmic properties in patients with hypertrophy.11,12 Catheter ablation techniques carry a paucity of data for HCM patients, yet remain an option for those patients who remain poorly controlled on drug therapy. 12
Thromboembolism risk-reduction therapy for AF patients is traditionally chosen based on the presence of a well-known set of risk factors (age, diabetes, vascular disease, female gender, a history of prior thromboembolic event, congestive heart failure, and hypertension). However, HCM patients, tending to be younger than the other high-risk groups, have not been included in the cohorts used to derive the traditional risk factors and carry a higher risk of thromboembolic events at 3.8% annual risk. 79 Thus, both the European Society of Cardiology (ESC) and ACC/AHA recommend that all patients with HCM and persistent or paroxysmal AF be treated with oral anticoagulation (OAC).11,12 For patients who are unable to take OAC, antiplatelet agents (aspirin or clopidogrel) can be used, though observational studies indicate that they are inferior for stroke prophylaxis when compared to OAC. No data exist regarding the use of the new oral anticoagulant agents in HCM patients, though they can be considered for patients unable to achieve a therapeutic International Normalized Ratio (INR) on vitamin K antagonists. 12
Electrophysiologic Device Therapy
Defibrillator Therapy
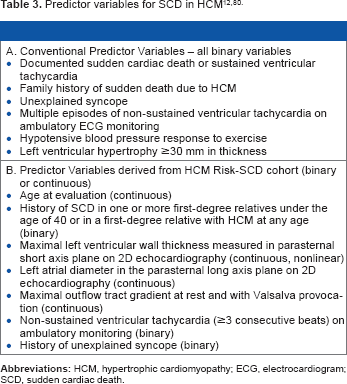
Defibrillator therapy is the only non-surgical therapy proven to reduce mortality in HCM patients when used in patients with appropriate risk. 14 The risk of SCD is not homogenous in the HCM population, and identifying patients at elevated risk remains a primary focus of HCM care.
Dual-chamber pacing
Dual-chamber pacing was postulated to provide benefit to patients with HCM for the same reason it was purported to worsen the health condition of patients with reduced LV function. Namely, the induction of dysynchronous ventricular contraction, which impairs stroke volume in patients with HFrEF, was shown to reduce outflow tract gradients in selected patients with HCM who are intolerant or refractory to medical therapy. 83 Initially viewed with great promise, subsequent randomized, double-blind, crossover trials showed no actual benefit. 84 In fact, the recent long-term follow-up data have suggested that continued dual-chamber pacing may have a deleterious effect on survival and incident heart failure when compared to the conventional therapy. 85
Biventricular Pacing
The role of biventricular pacing is less defined in the HCM population. There are emerging data that the use of biventricular pacing may reduce outflow tract gradients in patients with HCM who do not meet conventional resynchronization criteria (at least NYHA class III, LVEF <35% and QRS >120 ms).86–88 Additionally, small pilot studies have demonstrated an improvement in NYHA functional class, exercise time, and peak oxygen consumption with LV or biventricular pacing. 89 A biventricular pacing approach was also associated with a progressive and significant reduction in LV mass, most notably in the interventricular septum. 90
Septal-reduction techniques: alcohol septal ablation versus surgical myectomy
Despite optimal medical management, some HCM patients remain symptomatic with significantly elevated outflow obstruction (gradient >50 mmHg) either at rest or with exercise. Patients with persistent LV outflow tract obstruction, with symptoms (angina, syncope, or dyspnea), and without improvement with optimal medical therapy should be considered for septal-reduction intervention.11,12 Currently, two strategies exist for septal reduction: septal ablation and surgical myectomy.
In percutaneous septal ablation, absolute alcohol is selectively infused into the first or largest septal perforating branch off the left anterior descending artery. This serves to create a transmural infarct in the septum, thereby reducing the contractility, and eventually thickness of the proximal interventricular septum. Surgical myectomy involves the surgical resection of the hypertrophied septum to enlarge the outflow tract and improve gradients.
Debate still exists as to which strategy is superior. 91 It is becoming increasingly clear that, at centers having experience in myectomy, the surgical approach carries distinct advantages to the percutaneous approach in patients who can tolerate surgery. On average, myectomy lowers the outflow tract gradient to a greater degree than alcohol septal ablation.92,93 Patients undergoing successful surgical myectomy have a similar survival as the general population. 94 Septal ablation carries an increased risk of ventricular arrhythmias and an approximately 10-20% risk of need for permanent pacemaker because of heart block. 54 At experienced centers, surgical myectomy carries less than a 1% operative mortality risk. 94 Given these data, surgical myectomy is the treatment of choice for patients requiring septal-reduction intervention. 14 Alcohol septal ablation is listed as an alternative for those patients who carry an elevated surgical risk because of comorbidities or advanced age, or who are unconditionally averse to surgery.
Often, mitral valvular and papillary muscle abnormalities are complicit with the hypertrophied septum in contributing to outflow obstruction and mitral regurgitation. Mitral valve surgery is required in 10-20% of patients undergoing surgical myectomy. 95 Mitral valve replacement, posterior–superior realignment or partial excision with mobilization of the papillary muscles, and anterior mitral leaflet plication or extension have all been performed with varying degrees of success at the time of septal reduction.96–99 The presence of an elongated anterior mitral leaflet favors surgical repair over replacement. 100 A small case series of six patients with HCM undergoing percutaneous mitral valve repair with the Mitra-Clip device suggested that this approach was safe. However, two of the six patients had recurrence of their mitral regurgitation within one year of follow-up. 101
Advanced heart failure therapies
LV assist device (LVAD)
The use of a LVAD in patients with HCM is largely unexplored. In general, the small LV cavity size is not favorable for the current generation of continuous-flow LVADs because of the high risk of suction events and related arrhythmias. For this reason, patients with HFpEF are generally excluded from consideration of LVAD support. 102
However, two separate recent case series indicate that in carefully selected patients with HCM, the use of continuous-flow LVADs may be considered. In the first case series, four HCM patients were implanted with the HeartMate II® (Thoratec Corporation) axial flow device with selective operative myectomy enlarging the LV cavity size to accommodate the inflow cannula. These patients showed a prolonged survival when compared to HCM patients awaiting transplantation and undergoing standard care. 103 In the second report, 104 three HCM patients received the HeartWare® (HeartWare Corporation) centrifugal flow LVAD without concomitant myectomy. Despite having notably smaller LV size, the HCM patients achieved similar LVAD flows and had similar medium-term outcomes. One patient had ventricular inlet obstruction because of thrombus, one survived because of transplant after 708 days of support, and one was still on LVAD support (744 days) at the time of the publication.
Despite these selected reports, LVAD implantation in the general population of patients with HFpEF should only be considered in the highly selected end-stage patient.
Heart Transplantation
Heart transplantation is a therapeutic option for patients with HCM and refractory heart failure or life-threatening arrhythmias, though HCM patients compose a very small subset (~1%) of these patients in the United States. 105 Studies show similar outcomes for HCM patients and dilated cardiomyopathy patients undergoing heart transplantation. 106 Value cutoffs have been established for peak oxygen consumption and minute ventilation – carbon dioxide production relationship (VE/VCO2 slope) was measured during cardiopulmonary exercise testing as a prognostic indicator for patients with dilated cardiomyopathy to assess appropriate timing for transplant evaluation. While peak oxygen consumption has been shown to correlate with NYHA functional class, specific prognostic cutoff values have not been established in HCM patients.
Future Directions
Despite the great deal of scientific energy poured into the development of therapeutic options for patients with HCM and heart failure, many novel strategies are certain to be discovered in the future. Indeed, even the use of traditional heart failure therapies remains largely unexplored in any meaningful clinical trial for patients with HCM. For example, one can imagine the promise of antiremodeling agents such as ACE-inhibitors and ARBs in forestalling the progression of patients with genotype-positive, preclinical disease. In addition, several promising new diagnostic techniques and pharmacologic therapies for HCM are currently being studied.
Biomarkers
Galectin-3, an emerging prognostic biomarker in patients with heart failure, appears to be a mediator of increased myocardial fibrosis and a predictor of adverse clinical outcomes. 107 No studies exist to date investigating the clinical role of galectin-3 in patients with HCM, though with the pathophysiologic import that fibrosis plays in HCM, one might postulate that galectin-3 would be a particularly useful biomarker in HCM patients.
Ranolazine
Recently, studies have shown an impressive imbalance in electromechanical signaling in myocytes of patients with HCM, specifically enhancement of late sodium currents. 108 Ranolazine, a selective late sodium current inhibitor, has been shown to reduce angina and increase functional capacity in patients with coronary artery disease and anginal symptoms refractory to standard management. It carries the added physiologic benefit of improving myocyte diastolic function through the modulation of calcium sensitivity.109,110 All of these findings make ranolazine an attractive therapeutic option in patients with HCM, though no clinical studies have been completed to date demonstrating either safety or efficacy in this population.
Ivabradine
A novel funny channel blocker ivabradine has been shown to be beneficial in selected patients with coronary angina 111 and in patients with chronic heart failure. 112 While both of these findings require validation, the postulated mechanism of ivabradine and its ability to allow prolonged filling of the LV and coronary reservoir during diastole make it an intriguing therapeutic option in patients with HCM and outflow obstruction, anginal symptoms, and/or heart failure. 113 To date, only animal data are available with a feline model of HCM showing improved diastolic function with the administration of ivabradine.
Perhexiline
Cardiomycocytes in patients with HCM demonstrate a deranged metabolomic profile with abnormal energetics and inefficient energy handling. 108 Perhexiline, a metabolic modulator inhibiting the metabolism of free fatty acids and enhancing the use of carbohydrates by the cardiomyocyte, was recently studied in 46 patients with non-obstructive HCM. Intriguingly, perhexiline improved the metabolic profile of the LV and resulted in improved diastolic function and exercise tolerance. 114 A further study is needed, but these findings provide hope for cardiometabolics as a novel therapeutic target in HCM patients.
Conclusion
HCM is a worldwide cardiovascular disease. From its first description by Teare, Braunwald, and others 50 years ago, our current body of knowledge has rapidly advanced. We now understand that HCM represents a model of a monogenic, familial cardiomyopathy with incomplete penetrance and variable interindividual phenotypic expression. Refinements in imaging techniques such as echocardiography and cardiac MRI have provided unique insights into the pathophysiology of HCM and allowed for more precise diagnosis. Genetic testing now provides affected families with screening options. Likewise, advances in device therapy have provided many patients with the life-saving option of implantable defibrillator therapy, and improvements in surgical and percutaneous techniques have provided patients with septal-reduction therapeutic options.
Despite this rapid growth in scientific and clinical knowledge, much is left to discover about HCM. It remains woefully underrecognized in clinical practice. Despite our decades of experience, targeted pharmacologic therapeutic options are scarce, the patient selection for septal-reduction therapy (and modality of approach) remains controversial, and the utility of biventricular pacing is unknown. However, the coming years are sure to see advancements on all fronts. A further study into the genetic underpinnings of the disease may reveal fresh insight into its molecular pathophysiology. Novel pharmacologic agents targeting altered electromechanical signaling or deranged metabolomics have shown promise, and new biomarkers may provide an improved mechanism for prognostication or therapeutic targets. It is hopeful that we will continue to advance our scientific and clinical knowledge of this disease as much as we can over the next 50 years.
Author Contributions
Wrote the first draft of the manuscript: BAH. Contributed to the writing of the manuscript: BAH, GRS. Agree with manuscript results and conclusions: BAH, GRS. Jointly developed the structure and arguments for the paper: BAH, GRS. Made critical revisions and approved final version: GRS. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgment
The authors wish to thank Theodore Abraham, MD, for his assistance with Figures 2 and ![]() .
.