
Abstract
Periosteal new bone formation (PNBF) is the means by which appositional bone growth normally takes place on the surfaces of compact bone. Alterations in the periosteal microenvironment trigger complex interactions between osteoblasts and endothelial cells to promote PNBF. Physiologic processes like mechanical stress result in normal PNBF; but, a variety of pathologic processes result in excessive PNBF. The production of sufficient bone to be detectable by conventional radiography is a common feature of diverse etiologies, including infection; inflammation; prostaglandin E2 administration for ductal-dependent congenital heart disease; metabolic and hormonal abnormalities; neoplasms; fracture repair; systemic hypoxia; and hypertrophic osteoarthropathy. While the clinical settings and distribution of affected bone sites in these conditions are different, the histopathology of the PNBF is essentially identical; so, it seems logical that a common pathway might mediate them all. By combining the observations and insights gained from osseous research and studying the clinical pathology of these diverse conditions, we constructed a comprehensive pathway to explain PNBF. In doing so, it seems likely that Vascular Endothelial Growth Factor (VEGF) is the most likely common mediator of the pathways that lead to PNBF.
Introduction
Periosteal new bone formation (PNBF) is the means by which appositional bone growth normally takes place on the surfaces of compact bone. Alterations in the periosteal microenvironment trigger complex interactions between osteoblasts and endothelial cells to promote PNBF. Physiologic processes like mechanical stress result in normal PNBF; but, several pathologic processes result in excessive PNBF. The production of sufficient bone to be detectable by conventional radiography is a common feature of diverse etiologies, including infection; inflammation; prostaglandin (PG) E2 administration for ductal-dependent congenital heart disease (CHD); metabolic and hormonal abnormalities; neoplasms; fracture repair; systemic hypoxia; and hypertrophic osteoarthropathy (HOA). While the clinical settings and distribution of bone in these conditions are different, the PNBF histopathology is essentially identical; thus, it is plausible that a common pathway mediates them all. By combining the observations and insights gained from osseous research and studying the clinical pathology of these diverse conditions, we constructed a comprehensive pathway, the common mediator of which is Vascular Endothelial Growth Factor (VEGF). The production and effects of VEGF appear to explain the common endpoint of PNBF seen in myriad conditions.
An alphabetized list of abbreviations used commonly throughout the text is provided in Table 1.
Alphabetized list of abbreviations commonly used in text.

Overview of the Normal Periosteum
Structural Properties of the Periosteum
The periosteum is a thin fibrous layer on the surface of compact bone. It is loosely attached to the outer cortex by means of delicate, direct continuations of collagenous fibers, the Sharpey fibers (Bullough, 2004). In contrast, periosteal attachment to the cortical surface at the level of the ring of Ranvier, which surrounds the metaphysis at the level of the growth plate, is tight (Fig. 1a).
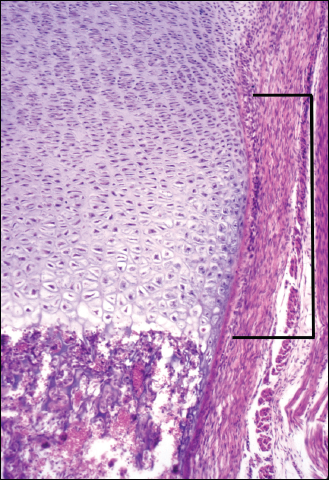
The ring of Ranvier, the site of periosteal attachment to the metadiaphysis, is illustrated by the bracket. (Routine hematoxylin-eosin stain, original magnification 90X).
The periosteum is physiologically and histologically subdivided into an outer fibrous layer (rich in fibroblasts, collagen, and elastin fibers) and an inner, more cellular cambium layer, named for its analogous position and homologous function in long bones to the growth layer on the surface of tree trunks. The cambium layer is the source of mesenchymal stem cells, osteoprogenitor cells, and osteoblasts necessary for appositional bone growth (Mills, 2007). When stimulated, periosteal mesenchymal stem cells become osteoprogenitor cells, which, in turn, differentiate into osteoblasts. Osteoblastic differentiation is characterized by early expression of alkaline phosphatase and later expression of DNA-binding transcription factor Runx-2/Cbfa-1 (vide infra). Runx-2/Cbfa-1 drives osteoblastic production of the collagen (Wagner and Karsenty, 2001) that constitutes osteoid (unmineralized bone). Mineralization of osteoid forms the new bone on the inner aspect of the periosteum adjacent to the cortex. This process is histologically identical to classic intramembranous (IM) ossification of the calvarium.
New periosteal bone may have a random, woven collagen fiber pattern or a more organized, lamellar collagen fiber pattern, depending upon its rate of production and maturation. With sufficient time and mechanical stress, osteoclasts remove immature woven bone and osteoblasts slowly replace it with mature lamellar bone. As the periosteal osteoblasts deposit new bone on the outer cortical surface, histiocyte-derived osteoclasts from the bone marrow slowly resorb the endosteal surface of the original cortex (Mills, 2007). These co-existing processes of bone apposition and resorption tend to limit cortical thickness while maintaining an endosteal space that is proportionate to bone diameter.
The cortex has an extensive collateralized vascular supply. The outer third of the circulation is derived from periosteal arteries that perforate the cortex and deliver blood to Volkmann canals and thence to Haversian systems. The endosteal nutrient arteries that furnish the circulation to the medullary cavity also supply the circulation to the Volkmann canals of the inner two thirds of the cortex. Anastomoses between the inner and outer circulation are primarily through Volkmann canals. This arrangement serves to maintain a dynamic intracortical microenvironment rich in oxygen, nutrients, and biochemical mediators. Unlike chondrocytes and fibroblasts whose viabilities are not impeded by an ossified matrix, osteocytes require close association with a blood supply and have limited transfer of oxygen and metabolites within bone (Mishra and Knothe Tate, 2003). This dependency imposes limits on the diameter of osteons in a Haversian system to approximately six concentrically arranged annular lamellae (or a distance of 200–250 μm from any given capillary) (Ham, 1952).
Age-and Site-Related Differences in the Periosteum and Physiologic PNBF
Numerous age- and site-related differences in the periosteum and physiologic PNBF have been identified. Compared with adult periosteum, late fetal and infant periosteum possesses a more cellular cambium layer containing numerous plump mesenchymal and osteoprogenitor cells (Figs. 1b and 1c). Moreover, immature periosteum is more richly vascularized than adult periosteum (Allen et al. 2004) and is less tightly adherent to the compact bone (Bullough, 2004) (Klein, 2005). In utero, this loose periosteal attachment may facilitate osteoblastic responses to mechanical forces exerted on fetal long bones by the uterine wall and developing ligamentous and tendinous attachments. Animal studies suggest that osteoblast responses to strain may require estrogen receptor (ER)-α activity (Lee et al. 2003). The estrogen-rich environment of pregnancy likely contributes to enhanced new bone formation by fetal periosteum (whereas relative estrogen deficiency of the post-menopausal state likely contributes to diminished new bone formation).

Histologic section of long bone periosteum closely applied to the underlying, mature cortical bone of the human adult. Note the thin, dense, outer fibrous, collagenous layer that blends almost imperceptibly with an inner, hypocellular cambium layer composed of small, scattered, spindle-shaped cells. (Routine hematoxylin-eosin stain, original magnification 250X).

Compared with adult periosteum, infant periosteum, as depicted in this illustration, shows a thicker and looser fibrous layer with numerous fibroblast nuclei visible within the collagen and elastin. This layer overlies a more cellular cambium layer that contains easily identified plump mesenchymal and osteoprogenitor cells. Immature woven bone of the cortex underlies the periosteum. (Routine hematoxylin-eosin stain, original magnification 250X).
The relatively immature composition and enhanced responsiveness of fetal periosteum continues in infancy. In term infants, the prevalence of PNBF in at least one long bone has been documented to range from 27% to 46% and as much as 78% in premature infants. Physiologic PNBF in infants is thought to represent a rapid period of bone growth via IM ossification resulting in a “double cortical layer” that gradually gets incorporated into the underlying cortex (Kwon et al. 2002).
During the infantile period, prior to weight-bearing activities such as crawling and walking, the high cellular activity and lax periosteal diaphyseal attachments facilitate appositional periosteal bone deposition. In infancy and childhood, as bone elongation occurs at the growth plates, tight periosteal attachments at the opposing rings of Ranvier exert tension on the periosteum in much the same way that a bow imparts tension to a bowstring. Since the periosteum is loosely attached to its underlying cortex, periosteal tension tugs the periosteum away from the underlying cortical surfaces. This periosteal separation, in turn, creates the electromechanical stresses that promote new bone formation by the cambium layer. Since periosteal tension is related to the underlying bone length, cortical thickness tends to remain proportionate to bone length. In normal ambulating children, the combination of gravity, ligamentous and tendinous attachments, and long bone growth provide the stimuli for physiologic PNBF. Appositional bone growth accelerates in the pubertal years due to hormonal stimulation.
As individuals age, Sharpey fibers of the periosteum become more tightly attached to the cortex (Klein, 2005). At the same time, cellularity, cell size, and vascularity diminish within the periosteal cambium layer. Correlatively, in vitro studies by Pfeilschifter et al. (Pfeilschifter et al. 1993) have shown that human bone cellular responsiveness to mitogenic hormonal and growth factors decreases with age. However, the adult periosteum, while diminished in cellularity and relative thickness, retains its bone-forming potential. This underlying capacity contributes to the periosteal reactions associated with trauma, infections, tumors, and other pathologic states. Although the rate of PNBF slows in the adult years, it tends to remain relatively more active in the short tubular bones of the hands and feet (personal observations, MJK) and likely accounts for localized PNBF seen in HOA (Fig. 2). Finally, the unique location of periosteal osteoblasts, in contrast to the endosteal cells of the inner cortex, may give these cells greater exposure to mechanical and biochemical stimuli (Allen et al. 2004).

Hypertrophic osteoarthropathy (secondary) in an adult male with lung carcinoma. Note the continuous layer of new bone apposed to the surfaces of the radius and ulna. (Contributed by Michael J. Pitt, M.D., Chief, Section of Musculoskeletal Radiology, The University of Alabama at Birmingham).
Periosteal cellularity varies with site along a given bone. Allen et al. (Allen and Burr, 2005) studied cadaveric bone samples from males and females ages 34–88 years and found that the periosteum of femoral neck was significantly less cellular than age-matched periosteum from the femoral diaphyses. This site-specific difference is less apparent in preterm and term infants (personal observations, OF-P).
Molecular Determinants of Normal Bone Formation and Remodeling and Associated Pathologic Conditions
Many factors contribute to bone formation and remodeling. Some factors were originally identified in investigations of skeletal growth and metabolism [i.e. bone morphogenetic proteins (BMPs)] (Wozney et al. 1988); however, subsequent research has demonstrated that several are later re-deployed in the postnatal period during times of stress and/or injury (Ripamonti, 2006). In the remaining text, we examine several of the key determinants in PNBF and attempt to demonstrate how VEGF is the apparent pivotal factor for all pathways resulting in radiologically-detectable excessive PNBF regardless of patient age or etiology initiating the process. A more complete list of molecular determinants of bone formation and remodeling (and likely PNBF) can be found in Table 2.
Alphabetized List of Molecular Determinants of Bone Remodeling.

Modified from Tables 1.3 and 1.4 from P. Bullough, Orthopaedic Pathology, 4th ed. 2004 (Ref 11).
Prostaglandin-Cyclooxygenase Pathways
PGs are best known for their role as potent mediators of inflammation; however, they are also stimulators and inhibitors of bone metabolism (Raisz, 2005). Their synthesis is initiated by cyclooxygenase (COX) isoforms, COX-1 and COX-2, which metabolize arachidonic acid to PGH2. Upon activation, specific synthases make PGD2, PGE2, PGF2, PGI2, and thromboxane A2 (Tilley et al. 2001). PGE2 is the main PG produced by bone cells; it has been shown to play a pivotal role in bone formation (Raisz, 2005) and fracture repair (Lee et al. 2007). Nonsteroidal anti-inflammatory drugs (NSAIDs), by targeting COXs, have been shown to inhibit fracture repair in animal models (Zhang et al. 2002).
PGE2 exerts its effects through four receptor subtypes, EP1R-EP4R (Tilley et al. 2001). EP2R and EP4R have been shown to play a role in the stimulation of bone formation and resorption by increasing cAMP-dependent activation of protein kinase A (PKA). Cyclic AMP and PKA production provides positive feedback to induce further COX-2 expression in osteoblastic cells and to amplify the inciting PGE signal (Sakuma et al. 2004).
Zhang et al. (Zhang et al. 2002) have shown that COX-2 regulates the induction of Runx-2/Cbfa-1 and osterix, factors critical for normal IM and endochondral bone formation during bone repair. Runx-2/Cbfa-1 is a DNA-binding transcription factor specific for osteogenic cells. It is a target of transforming growth factor (TGF)-β (vide infra) and BMP (vide infra); it regulates osteoblast differentiation (Ducy et al. 1997). Runx-2/Cbfa-1 is thought to play a key role in driving mesenchymal stem cells toward the osteoblast lineage and controlling bone formation by regulating the expression of osteoblast-specific marker genes (Zhang et al. 2002), (Janssens et al. 2005). Osterix acts downstream of Runx-2/Cbfa-1 and is essential for osteoblast differentiation and bone formation (Nakashima et al. 2002). It is thought to work together with endothelium-produced endothelin-1 to temporally and spatially link angiogenesis with bone formation and resorption (Qu and von Schroeder, 2006).
Conditions with elevated PGE and COX levels/activity and PNBF
Multiple physiologic and pathologic conditions with PNBF have associated increased PGE levels. Localized increased PGE levels have been found at sites of mechanical stress in vitro. Systemically increased PGE levels have been noted in clinical studies of children with Caffey's disease and in patients with certain infections. The various conditions are addressed, herein.
Mechanical stress
When mechanical forces act on bone, new bone is formed. Osteoblasts are the mechanosensory cells in bone; they demonstrate dose- and duration-dependent sensitivity to mechanical loading (Searby et al. 2005). Gap junctions are membrane channels that allow for cell-to-cell signaling (Li et al. 2006). Mechanically-induced connexin 43 hemichannel formation plays a major role in fluid flow shear stress-induced release of PGE2 (Jiang and Cherian, 2003). Physiologic levels of mechanical strain have been shown to induce periosteal cell proliferation and osteoblastic PGE2 production; endosteal osteoblasts have not demonstrated such response (Jones et al. 1991). Immature osteoblasts have been shown to be more sensitive than mature osteoblasts to mechanical loading stimuli (Searby et al. 2005). PGE2, via cAMP and PKA, translates mechanical loading stimuli into periosteal expansion via gap junction-mediated intercellular communication between osteocytes (Cherian et al. 2003).
Caffey's Disease(Infantile Cortical Hyperostosis)
Heyman et al. (Heyman et al. 1982) reported that PGE serum levels were elevated in children with Caffey's disease. This rare entity affects infants less than age six months and is believed to be of viral or immunologic etiology (Bullough, 2004). A genetic predisposition due to a missense mutation in the collagen type I alpha 1 (COL1A1) gene has been described. Infants with the disease develop soft tissue swelling and PNBF that primarily affects the long bones and mandible. Acute inflammation typically precedes the cortical thickening (Glorieux, 2005). Affected infants suffer unpredictable courses of disease but usually have spontaneous recovery within months (Bullough, 2004). Indomethacin, a nonspecific COX inhibitor, has been used to improve patients’ signs and symptoms (Heyman et al. 1982), (Glorieux, 2005).
Pge Administration in Ductus-Dependent CHD
Intravenous administration of PGE1, given to maintain patency of the ductus arteriosus in neonates with ductus-dependent CHD, is complicated by reversible and dose-dependent symmetrical PNBF (Fig. 3a) (Faye-Petersen et al. 1996). Ueda et al. (Ueda et al. 1980) first described cortical hyperostosis following long-term administration of PGE1 in two term infants with cyanotic CHD; bony changes regressed after cessation of therapy. In 1996 Faye-Petersen et al. (Faye-Petersen et al. 1996) compared postmortem bone radiographs and correlative bone histopathologies of three term infants with CHD who received varying durations of long-term PGE1 administration. While a spectrum of severity was noted, histologic examination of the bone showed abundant PNBF with a rich neovascular network whose branches were orthogonally oriented to the bone surface. The authors suggested that PGE1, and possibly cAMP, played a role in the neovascularization and that this neovascularity subsequently contributed to the formation of new bone and the observed bone hypertrophy. Figure 3b shows the light microscopic features of pathologic PNBF associated with long term PGE1 administration; but, as stated above, this histopathology is indistinguishable from that due to other causes.

PNBF in a three-month old female infant with ductal-dependent Transposition of the Great Arteries who received 50 days of intravenous PGE2 administration. There is marked expansion of the periosteum and so-called “bone within bone formation,” with the original cortex lying deep to periosteal new bone.

Light microscopically, pathologic PNBF consists of a cellular periosteal cambium layer overlying differentiating, long trabeculae of new woven bone in a vascular and edematous stroma. Newer trabeculae of PNBF lie immediately underneath the periosteum; the older bone is deeper and thicker. Capillaries branch at right angles from the periosteum and provide the critical network of oxygen and nutritional support to the osteoblasts and osteocytes of the growing bony trabeculae. (Routine hematoxylin-eosin stain, original magnification 200X).
Infections
PNBF has long been associated with infections, especially those occurring in utero. The classic example is congenital syphilis with associated Treponema pallidum vasculitis (inflammation of the vasa vasorum and accompanying vascular mural inflammation and destruction). Typical bony manifestations of congenital syphilis include destructive changes in the vomer; mandibular and maxillary bony alveoli and teeth; and “saber shin” malformations, consisting of a marked periostitis and cortical thickening that is most pronounced along the anterior aspect of the tibiae. In 1981 Oppenheimer et al. (Oppenheimer and Dahms, 1981) described the histopathologic bone findings in syphilitic infection as “periosteal new bone formation surrounding the older subperiosteal bone, with resultant periosteal elevation and thickening.”
Congenital cytomegalovirus (CMV) infection has been associated with pathologic PNBF referred to as “celery stalk” lesions on radiographs (Wigglesworth and Singer, 1991). Recent research has shown increased COX-2 mRNA, protein, and enzyme activity after infection of human fibroblasts with CMV. PGE2 levels have been shown to be transiently elevated in fibroblast cultures exposed to CMV; researchers have demonstrated that elevated levels of PGE2 are required for CMV to efficiently replicate in fibroblasts. COX-2 inhibition has been shown to eliminate PGE2 accumulation after CMV infection (Zhu et al. 2002), (Mocarski, 2002).
Herpes simplex virus (HSV) replication has also been shown to be enhanced by COX-2 activity and increased PG synthesis in cell culture and animal models (Mocarski, 2002).
Hormonal and Metabolic Factors
Multiple hormonal and metabolic factors play key roles in PNBF, including sex steroids, parathyroid hormone (PTH), vitamin A, and 1,25-dihydroxyvitamin D3 [1,25-(OH)2D3].
Sex Steroids
Estrogen and androgen receptors affect osteoblastic differentiation and function with ER-α activity (Lee et al. 2003) being the main mediator the bone protective effects of estradiol in both males and females. ER-α is highly expressed in periosteal osteoblasts and osteocytes (Bord et al. 2001) and is a major regulator of periosteal bone apposition (Allen et al. 2004). In vitro and in vivo studies have shown that osteoblastic stress-induced periosteal apposition is dependent on ER-α activity. Animal studies suggest that estrogens may increase the sensitivity of bone to mechanical stress (Lee et al. 2003). Vanderschueren et al. (Vanderschueren et al. 2006) showed that the effects of estrogen on PNBF are biphasic and concentration-dependent. In males and females, androgens stimulate PNBF. Low estrogen levels increase periosteal mechanical sensitivity; higher estrogen levels inhibit PNBF and/or its interaction with mechanical loading. Several authors attribute estrogen's inconsistent role in PNBF to its varying effects on its receptors (α and β) as well its varying effects on local cytokines and growth factors, such as interleukin (IL)-1, tumor necrosis factor (TNF)-α, growth hormone, and/or insulinlike growth factor (IGF)-I (Vanderschueren et al. 2006), (Raisz, 2005). Decreased estrogen levels in post-menopausal women may contribute to bone loss due to decreased ER-α activity and diminished responsiveness to mechanical stimulation (Lee et al. 2003). Post-menopausal women, despite bone loss, maintain some bone strength due to increased periosteal apposition (Ahlborg et al. 2003).
Pth, Vitamin a, and 1,25-Dihydroxyvitamin D3 [1,25-(oh)2D3]
PTH and PTH-related protein are important regulators of osteoblast function. In osteoblasts, they signal via the cAMP pathway (Said Ahmed et al. 2000) to induce the temporal expression of the proto-oncogene cfos during osteoblastic differentiation (McCauley et al. 1997).
Vitamin A (or retinol) levels influence bone formation, re-modeling, and metabolism. Vitamin A is present in food sources such as liver, kidney, and milk; the provitamin beta-carotene is found in plants (Lips, 2003). Chronic toxicity develops with prolonged excessive intake (Potts, 2005) and is associated with PNBF as demonstrated by hyperostoses in metacarpals, metatarsals, and other tubular bones such as the ulnae, tibiae, and fibulae. The characteristic histologic feature of chronic vitamin A toxicity is “periosteal apposition of coarse woven bone with large osteocyte lacunae” (Lips, 2003). Interestingly and despite lack of better understanding of role of vitamin A in PNBF, investigators have documented similar periosteal changes in patients treated with systemic retinoids (Brecher and Orlow, 2003) and in AIDS patients treated with the HIV protease inhibitor indinavir due to its possible interaction with retinoic acid signaling (Lenhard et al. 2000). Researchers have shown that retinoic acid and BMP (vide infra) cooperate to promote osteoblast differentiation (Skillington et al. 2002).
1,25-(OH)2D3 (or calcitriol) is a critical modulator of calcium metabolism shown to have anabolic effects on osteoblasts. 1,25-(OH)2D3 enhances osteoblastic production of VEGF, which, in turn, stimulates VEGF receptor gene expression on endothelial cells. Endothelial cells subsequently enhance the proliferation and differentiation of osteoblasts by producing osteotropic growth factors; the VEGF/VEGFR system was suggested to be involved in both bone formation and remodeling in vivo (vide infra) (Wang et al. 1997).
PTH, 1,25-(OH)2D3, and PGE2 have been shown to directly interact at the cellular level. PGE2 is thought to mobilize calcium in a manner similar to that after treatment with PTH (Said Ahmed et al. 2000). Studies have demonstrated complex interactions between 1,25-(OH)2D3 and TGF-β and their effects on osteoblast-specific gene expression. Smad protein signaling (vide infra) may mediate their crosstalk (Gurlek and Kumar, 2001).
Periostin
Periostin (or osteoblast-specific factor 2) is a signal peptide secreted primarily, and possibly exclusively, in the periosteum of bone in vivo. It is found in preosteoblasts and is released into the extracellular matrix (ECM) (Allen et al. 2004). Periostin is thought to facilitate osteoblast cell adhesion required for differentiation (Litvin et al. 2004). Levels are transiently increased during osteoprogenitor proliferation (and abnormal osteoblast proliferation) (Allen et al. 2004). Overexpression of Twist, a basic helix-loop-helix transcription factor, has been shown to increase periostin expression; Twist is thought to bind to the periostin promoter in undifferentiated preosteoblasts and up-regulate periostin expression (Oshima et al. 2002). Periostin levels decline as osteoblastic differentiation progresses, and, as such, it is thought to act as a negative regulator of osteoblast differentiation. Periostin expression is negatively regulated by 1,25-(OH)2D3 and positively regulated by TGF-β (Allen et al. 2004). Various isoforms of periostin are developmentally regulated and involved in osteoblastic recruitment, proliferation, and differentiation in vitro (Litvin et al. 2004).
Cytokines and Growth Factors
Multiple cytokines and growth factors have been shown to play a role in osteoprogenitor cell proliferation, osteoprogenitor to osteoblastic differentiation, and osteoblastic PNBF. Key mediators in PNBF include leukemia inhibitory factor (LIF) and TNF-α, IGF, fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), the TGF-β superfamily (including TGF-βs and BMPs), and VEGF.
LIF
LIF is a cytokine shown to regulate cell growth and differentiation in many different tissues, including bone. Researchers have demonstrated that LIF enhances differentiation of preosteoblasts and acts directly on osteoblast function, producing dose-related increases in DNA and cells. Osteoblasts, in turn, have been shown to synthesize LIF in vitro. Osteoblastic production of LIF mRNA is increased by TNF-α with or without retinoic acid (Cornish et al. 1993).
IGF
IGF I and II, polypeptides structurally similar to insulin, are bound to and modulated by IGF binding proteins (IGFBPs). By IGF-dependent and IGF-independent mechanisms, IGFBPs are thought to help modulate bone cell proliferation (Hwa et al. 1999).
IGF-I, present in the bone matrix, is released during bone resorption. The cytokine is also produced by osteoblasts in response to PTH and growth factors (Goad et al. 1996). PGE2 and other cAMP-mediated factors have been shown to stimulate IGF-I synthesis in osteoblast-enriched cultures from fetal rat bone (McCarthy et al. 1991). IGF-I acts (via autocrine or paracrine mechanisms) on bone to stimulate the proliferation of preosteoblasts; it has been shown to promote bone matrix formation by differentiated osteoblasts. IGF-I is increased in fracture sites where it has been shown to induce VEGF mRNA and protein expression, thus facilitating the coupling of angiogenesis with bone repair (Goad et al. 1996). Using an animal model, Japanese researchers showed that IGF-I administration may play an important role in the early induction of bone formation from grafted periosteum (Ueno et al. 1999).
FGF
FGFs comprise a family of twenty-two genes and six subfamilies of proteins. Most members of the FGF family bind and signal via four FGF receptor tyrosine kinase molecules. FGFR1 and FGFR2 are thought to be the two primary receptors involved in IM bone formation.
FGF2 and FGF18 have been shown to be important for calvarial development. In vitro studies have shown that FGF2 may contribute to the regulation of IM calcification, while FGF18 is thought to regulate calvarial osteoblast differentiation. FGFs have been shown to regulate other key factors in bone formation, such as IGFs, IGFBP-5, TGF-β, and VEGF. Researchers suggest that FGFs may work together with BMP to control calvarial growth and differentiation during IM bone development. Interestingly, calvarial osteoblasts produce FGF2; this production is up-regulated by FGF2 itself, PTH, PGE2, and TGF-β (Ornitz and Marie, 2002).
PDGF
PDGF is a polypeptide found in platelets as well as other cell types, including bone. PDGF is the dimer product of two genes that encode PDGF chains, PDGF-A and PDGF-B, that subsequently form PDGF-AA, -BB, or -AB. PDGF is thought to be a regulator of cell growth. Unstimulated human and rat osteoblasts express the PDGF-A gene (and possibly the PDGF-B gene) (Hock and Canalis, 1994). A few authors have described the complex interplay between PDGF and IGF-I in regard to calvarial DNA synthesis and collagen metabolism (Hock and Canalis, 1994), (Canalis et al. 1989). Hanks et al. (Hanks et al. 1986) demonstrated how PDGF in extravasated blood at a site of injury may stimulate appositional bone formation beneath an intact periosteum.
Tgf-β Superfamily
The TGF-β superfamily consists of approximately thirty dimeric cytokines that are structurally-related. Two of the main superfamily members involved in bone formation are TGF-β and BMP.
TGF-β1 is the prototype of the TGF-β superfamily and is the most abundant isoform in bone. TGF-β1 has been shown to affect both bone formation and resorption and is thereby a key player in bony remodeling. In vitro, TGF-β1 promotes bone formation by recruiting osteoblast progenitors and stimulating their proliferation. It promotes early stages of osteoblastic differentiation; later stages are thought to be blocked by TGF-β1 and instead regulated by BMP and other factors.
TGF-β1 is made and secreted by bone cells as biologically inactive small latent complexes that consist of a latency-associated peptide and a mature peptide. Small latent complexes are stored in the ECM and become active mature TGF-β via osteoclastic activity. In this instance, bone resorption via osteoclasts is halted, and TGF-β1-induced osteoblast proliferation and differentation takes place. TGF-βs signal through Runx-2/Cbfa-1, R-Smads (i.e. R-Smads 2/3), and Smad-independent pathways to regulate gene transcription.
In vivo, the effects of TGF-β1 on osteoblasts and osteoclasts depend on cell differentiation, cell density, TGF-β1 concentration, and the presence of other growth factors in the bony microenvironment (Janssens et al. 2005).
BMPs also belong to the TGF-β superfamily; they are a group of structurally-related growth and differentiation factors that were originally identified in their ability to induce ectopic bone formation. Today, they are largely recognized as being the most important regulators in bone formation. Upon activation, BMPs trigger a cascade of events leading to osteoblast differentiation. In normal IM bone, BMPs 3, 4, 7, and 8 are highly expressed. When the bony microenvironment is altered; however, other BMPs may manifest (Suttapreyasri et al. 2006).
BMP-2 is expressed during murine embryonal skeletogenesis; it stimulates osteoblast differentiation from uncommitted progenitors both in vitro and in vivo (Chikazu et al. 2002). BMP-7 (or osteogenic protein) has been shown to stimulate new bone formation in vivo and to induce osteoblastic cell proliferation and differentiation in vitro (Yeh and Lee, 1999).
BMPs signal via three serine/threonine kinase receptors, BMPR-IA, BMPR-IB, and BMPR-II. BMP ligands bind to hetero-oligomeric complexes of BMPR-I and BMPR-II on the cell surface. Upon ligand binding to type-I and type-II BMPR complexes, BMPR-II phosphorylates BMPR-I. Phosphorylated active BMPR-I then phosphorylates receptor (R)-Smads 1/5/8. R-Smads 1/5/8 form heterodimers with Smad 4, a common-partner Smad. Together, the Smads translocate to the nucleus where they regulate the transcription of certain genes, such as alkaline phosphatase and distal less homeobox 5, a protein shown to play an important role in osteoblast differentiation. In vitro studies by Singhatanadgit et al. (Singhatanadgit et al. 2006) showed that growth factors involved in the proliferation and differentiation of osteoprogenitor cells, such as TGF-β1, FGF-2, and PDGF-AB, increased the expression of cell surface BMPR-IB thereby enhancing osteoblastic sensitivity to BMP ligands. Zaidi et al. (Zaidi et al. 2002) showed that Runx-2/ Cbfa-1 is required to target TGF-β and BMP-2-dependent Smads to subnuclear sites.
Separate authors have shown that PGE2, via binding of EP4 on preosteoblasts, enhances bone formation by inducing expression of Runx-2/Cbfa-1 and BMP-2 (Arikawa et al. 2004). BMP-2, in turn, transcriptionally induces COX-2 expression (and PGE2 production) in osteoblasts via a Runx-2/Cbfa-1 DNA-binding site (Chikazu et al. 2002).
Recombinant human BMP has been used experimentally and clinically to promote wound healing and regeneration processes in human bone (Singhatanadgit et al. 2006).
VEGF
Independent but converging avenues of research have lead to the identification and characterization of VEGF, a gene and protein having a vital role in angiogenesis, vascular permeability (Ferrara, 2004), and bone morphogenesis (Furumatsu et al. 2003). Members of the VEGF gene family include VEGF A-D, placental growth factor, and VEGF-like proteins of viral origin. PDGF is a VEGF homologue. VEGF-A is the prototype molecule and is hereafter referred to as VEGF (Ferrara et al. 2003).
VEGF is a 45 kDa heparin-binding homodimeric glycoprotein whose signaling is a critical rate-limiting step in physiologic angiogenesis; it is also considered a major regulator in pathologic angiogenesis (Ferrara, 2001). At least four human VEGF splice variants with varying heparin-binding capabilities are recognized. VEGF165 is the main isoform; it is both diffusible and bound to the endothelial cell surface and ECM (Ferrara, 2004). Heparin-binding forms of VEGF can release other angiogenic factors (such as FGF) stored in the ECM and synergize with VEGF to induce angiogenesis (Jonca et al. 1997), (Asahara et al. 1995).
VEGF has been shown to bind to two of the three VEGF tyrosine kinase receptors, VEGFR-1 (fms-like tyrosine kinase) and VEGFR-2 (kinase insert domain-containing receptor)/fetal liver kinase-1), both of which are expressed and have been shown to mediate its endothelial effects (Neufeld et al. 1999). Some authors propose that VEGFR-1 is the primary receptor in mediating VEGF's effects on primary human osteoblasts (Mayr-Wohlfart et al. 2002).
Hypoxia
Pathologic conditions, such as local and systemic hypoxia and HOA, have been shown to have elevated levels of VEGF and PNBF. Table 3 lists other disease conditions associated with radiographically-detected PNBF.
Other Conditions Associated with Radiologically-Detected PNBF.

Modified from Gamuts (Chapter IV) from H. Taybi and R. Lachman, Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasias, 4th ed. 1996 (Ref 78).
Local(Fracture Site)hypoxia
Changes in cellular oxygen tension can result in physiological and pathological consequences that alter gene expression (Faller, 1999). Bone injury results in disruption of blood vessels and vasoconstriction, thus making the fracture site a quintessential setting for studying the cellular effects of local hypoxia (Bouletreau et al. 2002). Fracture site hypoxia is thought to be a major determinant of all phases of bone repair (Street, 2005). The physiologic process of cortical bone repair at a fracture site occurs via IM ossification (Street et al. 2002); the vasculature of bone is an essential component of the process (Street, 2005).
Hypoxia is directly and indirectly angiogenic. It enhances human endothelial cell proliferation and blood vessel formation and stimulates VEGF release from primary human osteoblasts in vitro (Street, 2005), (Steinbrech et al. 2000). Hypoxia causes altered expression of vasoactive substances and matrix proteins that modulate vascular tone (upon short-term exposure); hypoxia participates in the remodeling of the vasculature and surrounding tissue (upon long-term exposure) (Faller, 1999). Hypoxia affects osteoblasts at the fracture site by regulating cell proliferation, cell migration, and ECM protein synthesis through the induction of cytokines and cell signaling pathways (Street, 2005). VEGF is thought to link bone growth and angiogenesis by mediating a paracrine interaction between osteoblasts and endothelial cells (Street, 2005).
Hypoxia acts indirectly on bone repair via the stabilization of the transcription factor hypoxia-inducible factor (HIF)-1. HIF-1 is a heterodimer with α and β subunits. The HIF-1α subunit is regulated by hypoxia. Under hypoxic conditions, HIF-1α escapes proteosomal degradation (McPherson and Pincus, 2007) and has been shown to activate many gene products, including VEGF, VEGFR-1, erythropoietin, and nitric oxide synthetase (Gerber et al. 1997). Steinbrech and al. (Steinbrech et al. 2000) proposed an oxygen-sensing mechanism (similar to erythropoietin) in the regulation of osteoblast VEGF expression. HIF is thought to bind to a VEGF transcription initiation site to induce VEGF transcription (Gerber et al. 1997). In vivo up-regulation of the VEGFR-2 is thought to be mediated by a paracrine factor released by ischemic tissues (Brogi et al. 1996) or by post transcriptional mechanisms such as increased VEGFR mRNA stability (Waltenberger et al. 1996). In addition to hypoxia, other factors induce HIFα stability and transcriptional activity. These factors include mechanical stress, cytokines, growth factors, hormones, microorganisms, and reactive oxygen species (Gaber et al. 2005).
VEGF is constitutively expressed by primary human osteoblasts in normal oxygen culture conditions. Levels have been shown to increase in a dose-dependent manner in hypoxic conditions (Steinbrech et al. 1999). Street et al. (Street, 2005) demonstrated that a reciprocating interaction exists between osteoblasts and endothelial cells and that this process is enhanced by hypoxia. When subject to short-term low oxygen tension, osteoblasts adapt by releasing VEGF. Hypoxic endothelial cells release osteogenic mitogens, including IGF-I.
The fracture microenvironment is rich in inflammatory mediators that are thought to act directly and indirectly in the bone repair process. TGF-β1 and BMP expression have been shown to be increased in fracture healing (Bouletreau et al. 2002), (Saadeh et al. 1999). The acidity and plasminogen of the fracture microenvironment are thought to activate and up-regulate TGF-β1, allowing it to stimulate osteoblast migration, to modulate osteoblast proliferation, and to regulate osteoblastic VEGF expression. Exogenous TGF-β1 application has been shown to accelerate closure of membranous bone defects (Saadeh et al. 1999).
Human microvascular endothelial mRNA expression of BMP-2 is increased in hypoxic conditions. Bouletreau et al. (Bouletreau et al. 2002) showed that human microvascular endothelial cells, when stimulated by hypoxia (or recombinant human VEGF), had up-regulation of BMP-2 mRNA hours after exposure. The effects of hypoxia and VEGF were additive on BMP-2 mRNA expression in bovine capillary endothelial cells. Zhang et al. (Zhang et al. 2002) suggested that COX-2 and PGE, also prevalent in the fracture callus, are required for the correct expression of Runx-2/Cbfa-1 and osterix. Zhang's model of bone injury and inflammation suggests that COX-2 is induced to produce increased amounts of PGE2. PGE2 then acts directly or, more likely, indirectly via VEGF with BMP-2, to increase Runx-2/Cbfa-1 and osterix. PNBF is presumed to follow.
Systemic hypoxia
Severe systemic hypoxemia produces a similar microenvironment to that seen in bone fractures. Patients with oxygen deprivation have decreased pulmonary metabolism of PGE (Pisarello et al. 1997). Extrapolating this data, it can be hypothesized that in infants with susceptible periosteum, systemic hypoxia due to prematurity, CHD, infection, or other disease conditions may contribute to increased levels of circulating PGE and to the observed bony consequences. Likewise, though to a lesser effect, hypoxemic adults would be expected to manifest similar bony changes.
HOA
HOA is characterized by periostitis followed by symmetrical PNBF along the distal ends of long bones. Other disease manifestations include clubbing of the digits, arthritis, and synovial effusions. HOA is thought to be secondary to distal ischemia and subsequent release of bioactive mediators, including VEGF and PDGF (Spicknall, 2005).
HOA is subdivided into primary and secondary forms. Primary HOA is rare familial condition with no identifiable etiology. It is more common in males and has a bimodal age distribution, occurring in the first year of life and, more commonly, at puberty. Prominent features include clubbing, painless perostitis, and skin thickening and increased coarseness. Secondary HOA is more common than primary HOA and is associated with painful, non-inflammatory joint effusions in adults. It occurs in 5–10% of patients with primary intrathoracic malignancies, notably bronchogenic carcinoma and pleural tumors. Secondary HOA additionally manifests in numerous other chronic disease processes (Gilliland, 2005). Treatment for secondary HOA entails treating the underlying disorder; symptom control has been reported with aspirin, other NSAIDs (Gilliland, 2005), and octreotide (a VEGF inhibitor) (Angel-Moreno Maroto et al. 2005).
Discussion
The disorders in infants, children, and adults in which PNBF is a common feature are diverse, but our multidisciplinary review of the literature of normal and abnormal bone formation and these disorders indicates that the diverse processes are all linked by a common pathway. Mechanical stress, Caffey's disease, iatrogenically-induced increased levels of PGE, infections, inflammation/acidosis, hypoxia, and HOA stimulate mediators, including COX-2, TGF-β1, BMP-2, HIF, hormones, and other cytokines and growth factors. These, in turn, induce osteoblastic release of VEGF, which stimulates angiogenesis and, via autocrine effects and increased endothelial cell expression of BMP-2, osteoprogenitor cell proliferation and differentiation. Previous reports have suggested that VEGF may act as a central mediator for other factors in coupling angiogenesis with bone formation and remodeling (Street et al. 2002), but, to our knowledge, none has pursued this relation nor explored the potential role of VEGF in the diverse spectrum we have examined. Figure 4 is the schematic synthesis of our comprehensive study, as discussed vide infra.

Diagram depicting a synthesis of the literature of the complex etiopathogenetic pathways involved in PNBF. The center of the diagram shows the normal path of differentiation of primitive periosteal mesenchymal cells to periosteal osteocytes of PNBF. The endpoints of cascades and/or sites of activity of most of the myriad molecular and chemical mediators and stimulating factors linked to bone formation, such as mechanical stress and local hypoxia, are shown on either side of this pathway. Also integrated in the diagram are sites where pathologic conditions or mediators feed into and alter the normal pathways involved in PNBF and thereby result in excessive pathologic new bone formation along the shafts of tubular bones. For example, PG production is revealed to be an initial key component in PNBF; PG is a mediator for the physiologic effects of mechanical stress as well as pathologic PNBF associated with systemic hypoxia, bacterial and viral infections, and iatrogenic and underlying disorders with elevated PGE levels. BMP-2 is revealed as a multisite stimulator and a positive feedback mediator in normal and pathologic PNBF and is involved in fracture-associated local hypoxia and systemic hypoxia. The interactions among the growth factors, chemical mediators, and hormones are discussed in detail in the text, but the interrelationship between FGF and IGF-1 is depicted, as an example. Finally, the diagram reveals that, whatever the initiating cause of PNBF, VEGF is likely the final, pivotal mediator required for bone formation; blood supply (angiogenesis) is requisite for osteoblast/osteocyte viability, and the production of VEGF by osteoblasts as well as endothelial cells ensures their longevity. Moreover, the positive feedback loop by VEGF on BMP-2 production amplifies the proliferation and differentiation of osteoprogenitor cells at all levels. Pathologic PNBF, illustrated at small scale at the bottom of the diagram (vide also Figure 3b), results from an imbalance in the factors that drive the proliferation of bone forming cells and ultimately, the production of VEGF. Conditions that elevate VEGF also result in pathologic PNBF. Figure graphics were provided by David Fisher, UAB Medical Education and Design Services.
The backbone of the graphical depiction establishes the role of BMP-2 in periosteal mesenchymal cell commitment to the osteoprogenitor lineage. BMP-2, together with TGF-β1, subsequently promotes osteoblastic differentiation and proliferation.
PGs and COXs have long been associated with PNBF. PGE levels are elevated in patients with Caffey's disease; patients have bony symptoms relieved with COX inhibitors. Neonates given intravenous forms of PGE1 to maintain patency of the ductus arteriosus in ductus-dependent CHD also demonstrate reversible and dose-dependent symmetrical PNBF. Patients with systemic hypoxia have decreased clearance and elevated levels of PGE. Bacterial and viral infections have been associated with PNBF, especially infants infected in utero, with periosteal, hormonal, and environmental factors likely synergistically contributing to the bony findings. CMV and HSV have been shown to be associated with elevated levels of circulating PGE2.
Mechanical stress also feeds into the schematic at the level of PGE. Physiologic levels of mechanical strain induce osteoblastic PGE2 production with PGE2 serving to translate mechanical loading into periosteal expansion via osteocytic gap junction-mediated intercellular communication. PGE2 exerts its effects by binding to EP2 and EP4 receptors on osteoblasts, causing increased cAMP and PKA production and further COX-2 release; more COX-2 perpetuates the cycle. Harada et al. (Harada et al. 1994) showed that PGE2 causes increased expression of VEGF mRNA and protein in osteoblastic cells via a cAMP-dependent PKA signaling pathway.
COX-2 maintains a population of mesenchymal cells in a preosteoblast state during normal conditions. With injury, increased COX-2 levels support osteoblastic differentiation. COX-2 may act via regulation of Runx-2/Cbfa-1 and osterix (Zhang et al. 2002). Chikazu et al. (Chikazu et al. 2002) showed that BMP-2 transcriptionally induces COX-2 in osteoblasts via the Runx-2/Cbfa-1 binding site.
Multiple additional factors / conditions ultimately enter the schematic at various stages during osteoprogenitor cell proliferation and differentiation to promote new bone formation. These include inflammation / acidosis, periostin, IGF-I, PDGF, LIF, sex steroids, PTH, FGF, vitamins A and D, and HIF.
In addition to the requisite mediators, in order for osteoblasts to form bone, the microenvironment must have adequate blood flow. An interdependent and complex relationship exists between osteoblasts and endothelial cells. Recent studies have explored this relationship both in vivo and in vitro. VEGF is thought to act on osteoblasts to induce osteoblastic proliferation and migration (Mayr-Wohlfart et al. 2002). VEGF expression by osteoblasts has been shown to be stimulated by hypoxia, PGE, IGF-I, FGF-2, TGF β1-3, BMPs-2, -4, and -6 (Furumatsu et al. 2003), and multiple other factors. VEGF levels are increased in HOA. VEGF stimulates endothelial cells to make new vessels. Endothelial cells release BMPs that further stimulate angiogenesis through osteoblastic VEGF production (Bouletreau et al. 2002). VEGF, as such, is thought to couple angiogenesis to bone formation (Deckers et al. 2000).
Multiple physiologic and disease entities and manifestations alter the local or systemic microenvironment such that VEGF mRNA and protein expression are up-regulated with excessive PNBF as a common endpoint.
Conclusion
Our comprehensive construct and distillation of the literature suggests that the generation of VEGF plays a pivotal role in PNBF. VEGF is secreted by osteoblasts and, via binding to VEGFR-1 receptors on osteoblasts, acts in an autocrine manner to induce osteoblastic proliferation and migration. VEGF indirectly induces proliferation and differentiation of osteoblasts by stimulating endothelial cells to produce osteoanabolic growth factors (Mayr-Wohlfart et al. 2002), such as BMPs. In its best-understood role, VEGF acts on endothelial cells to promote angiogenesis, an essential process for bone formation, remodeling, and fracture repair.
We propose that VEGF is the common denominator in the histo-radiographic findings of PNBF in infants, children, and adults with various disease conditions. These conditions trigger a plethora of factors which directly or indirectly stimulate VEGF in promoting angiogenesis, as well as periosteal mesenchymal and bone cell proliferation and differentiation necessary for new bone formation. Our pathway unifies the work of many investigators leading to a common mechanism for PNBF. VEGF appears to be the fulcrum in all normal and pathologic processes resulting in PNBF, regardless of patient age or inciting event.
Footnotes
Acknowledgements
This study was reviewed and fully approved by the authors’ Institutional Review Board.