
Abstract
Array-based comparative genomic hybridization (aCGH) allows measuring DNA copy number at the whole genome scale. In cancer studies, one may be interested in identifying DNA copy number aberrations (CNAs) associated with certain clinicopathological characteristics such as cancer metastasis. We proposed to define test regions based on copy number pattern profiles across multiple samples, using either smoothed log2-ratio or discrete data of copy number gain/loss calls. Association test performed on the refined test regions instead of the probes has improved power due to reduced number of tests. We also compared three types of measurement of copy number levels, normalized log2-ratio, smoothed log2-ratio, and copy number gain or loss calls in statistical hypothesis testing. The relative strengths and weaknesses of the proposed method were demonstrated using both simulation studies and real data analysis of a liver cancer study.
Introduction
DNA copy number is the number of copies of DNA at a genome region. Gain and/or loss of chromosomal regions have been associated with various human diseases including cancer. 1 Therefore detecting and mapping DNA copy number changes provide a systematic approach to understand the association between DNA copy number abnormality and human disease. DNA copy number changes in tumor tissues are often referred to as DNA copy number aberrations (CNAs) which vary in size from 1 Kb up to one complete chromosome arm. High resolution array-based comparative genomic hybridization (aCGH) allows measuring DNA copy number at hundreds of thousands of probes or locations throughout the genome.2,3
Different methods and algorithms have been developed to divide the genome into regions with the same copy number level for a single sample using diversity techniques such as Hidden Markov Models, 4 circular binary segmentation,5,6 mixture models, 7 and Bayesian change point analysis. 8 Recently research has been devoted to identify recurrent CNAs that are shared across multiple samples. This is an important issue because chromosome regions where recurrent CNAs occur may play important roles in the development and progression of cancer and other human diseases. Rouveirol et al 9 computed the recurrent minimal genomic alterations based on discretized CGH profiles. Shah et al 10 extended single sample HMMs to multiple samples to identify recurrent CNAs by jointly inferring CNA patterns and making gain/loss calls. Zhang et al 11 proposed simple scan and segmentation algorithms based on the sum of the chi-square statistics for each individual sample to give a sparse and intuitive cross-sample summary. Multiple-sample segmentation and detection of recurrent CNAs are still challenging research areas both computationally and conceptually. For more references, see the review paper. 12
In clinical cancer studies, clinicians and researchers may be interested in identifying CNAs associated with patients’ clinical outcomes and tumor pathological characteristics. Unfortunately, little research has devoted on this type of downstream analysis, which has more clinical significance and is different from identifying recurrent CNAs as in most of the previous studies. Moreover, some studies have performed downstream analysis on whole chromosome level or chromosome arm level. Due to a lack of fine mapping of chromosome regions, this approach may not be efficient if only some regions rather than complete arm of the chromosome are associated with the interested clinical events. On the other hand, if association test is performed for each probe, a large number of tests (hundreds of thousands up to millions) are inevitable. In addition, probes adjacent on chromosome are more likely to have the same copy number status and will result in highly correlated tests. Here, we proposed to define test regions based on DNA copy number pattern profiles across multiple samples, and perform association tests on these test regions instead of individual probes. Furthermore, excluding non-variant test regions would result in smaller number of tests and improved power. We also compared the use of normalized log-ratio, smoothed log-ratio, and discrete copy number gain/loss calls in downstream analysis. The relative strengths and weaknesses of these measurements and the proposed method were evaluated using simulation studies and real data analysis of a liver cancer study.
Methods
Measurements of copy number level
Normalized log2-ratio
In an aCGH experiment, the test sample and the reference sample are labelled with different dyes and co-hybridized on a microarray, which contains hundreds of thousands to millions of probes depending on the array platform. Usually, the test sample is labelled with Cy5 and the reference sample with Cy3. Log2-ratio of background-corrected Cy5 signal to background-corrected Cy3 signal is computed for each probe on the array. The resulting ratio at a probe provides an estimate to the ratio of copy numbers of the corresponding DNA sequences in the test and the reference samples. In general a normalization step is needed to remove systematic array effect.
Smoothed log2-ratio
A smoothing or segment detection method is applied to normalized log2-ratios to divide the genome into regions with the same copy number levels. Adjacent regions have different copy number levels. The segmented or smoothed log2-ratio is assigned to be the median or average of the normalized log2-ratios of the probes contained in that region.
Gain or loss calls
DNA copy number gain or loss calls are usually made based on the smoothed log2-ratios. For the regions where no copy number changes occur the smoothed log2-ratios are close to 0. When there are copy number gains (or losses), the smoothed log2-ratios are significantly greater (or less) than 0. However there are no hard thresholds on choosing the significance level to make gain or loss calls. Users decide whether there is sufficient evidence to call a probe having copy number gained or lost. For example, Aguirre et al 13 considered a segment having gain or loss if the corresponding smoothed log2-ratio is more than four standard deviations away from the middle 50% quantile of data; Willenbrock and Fridlyand 14 defined the threshold using three times median absolute deviation (MAD) of difference between observed and smoothed log2-ratios.
Defining test regions
When smoothed log2-ratios or gain/loss calls are used, we define “test regions” on which downstream analysis will be performed. Figure 1 shows DNA copy number profiles for three subjects on 90 contiguous probes. Each dot represents the normalized log2-ratio at a probe. The solid (red) lines represent the smoothed mean log2-ratios. There are three, two and two segments for subject 1, 2 and 3, respectively, in this genomic region. When the test regions are defined based on the smoothed log2-ratios, there are three test regions consisting of probes 1–30, 31–60, and 61–90, respectively. When gain/loss calls are used, test regions depend on what criterion is used. Suppose we consider copy number changed when the absolute value of smoothed log2-ratio is greater than 0.50, then there is no copy number change for probes 1 to 60, and there is copy number gain for probes 61–90, for subject 1. Thus there are two test regions consisting of probes 1–60, and 61–90, respectively. The set of probes in a test region remains constant within a sample and shares the same profile across samples, therefore the association tests for these probes will be exactly the same.

An example of DNA copy number profiles for three subjects on 90 consecutive probes. The gray dots are the raw log2-ratios of copy number measurements, the red straight lines represent the smoothed log2-ratios for the identified segments, and the vertical dashed black lines represent the test regions based on the smoothed log2-ratios.
We formally define a test region as a set of contiguous probes on the same chromosome in the following. Let

where |
Identifying non-variable test regions
The purpose of hypothesis testing discussed in this paper is to identify genome regions which are associated with certain clinicopathological characteristics; therefore non-variable test regions will be excluded from further analysis. For example, the test region containing probes 61–90 (Fig. 1) are recurrent CNAs which are an important characteristic for identifying disease-related biomarkers. With all subjects having the same copy number status, it indicates that this region is not associated with the interested clinical event and it will not be tested. Similarly the first test region (Fig. 1) where probes 1–30 have no copy number changes, is not of interest either and excluded from further analysis. When gain/loss calls are used to identify CNAs with clinical significance, usually only probes with CNA frequency greater than a threshold (such as > 10% as used in) 15 are included in the analysis for clinical application value.
When smoothed log2-ratios are used in the analysis, it is not obvious to identify non-variable test regions as using discrete gain/loss calls. When each biological sample has technical replicates, one can separate the variation between the samples from the measurement errors which will help us evaluate the variation of the probes across samples. When there is no technical replicate, one can compute the variation or standard deviation of the log2-ratios across samples by employing similar strategy as in microarray studies. If the variation is small, it is more likely that the corresponding region is a non-variable test region.
Performing association tests
After defining test regions and excluding non-variable ones, association tests are performed. In this paper, as an example, survival analysis using Cox proportional hazards model
16
for smoothed log2-ratios or log-rank test
17
for gain/loss calls was applied to identify CNAs associated with cancer metastasis. Let

and it has the following proportional hazards structure

where
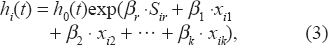
where
For each test region, the
Liver cancer data
The proposed methods will be applied to a liver cancer data set. Sixty-three newly diagnosed HCC patients who underwent radically surgical therapy at Eastern Hepatobiliary Surgery Hospital, Shanghai, China, from December 2007 to March 2008 were recruited. All patients were ethnic Han Chinese, and none had received radiation therapy or chemotherapy before surgery. A total of 63 tumor tissue samples and 11 matched surrounding non-tumor liver tissue samples were frozen in liquid nitrogen within 1 h after surgical removal and kept at –80 °C until DNA extraction. Final diagnoses were pathologically confirmed by pathologists from H&E-stained slides. Demographic data were obtained through in-person interviews at the first hospital admission. Information on tumor characteristics, such as tumor differentiation, envelope status, thrombus status and tumor stage, was made on the basis of pathological and medical reports. The 63 patients have been followed for 2.5 years on average after the surgery and the clinical events including distant metastasis were recorded. In total, 8 (12.7%) subjects developed distant metastasis during the follow-up, among which 7 subjects developed lung metastasis and 1 developed bone metastasis. Diagnosis and presence of distant metastases were based on: (1) positive findings on cytological or pathological examination, and/or (2) positive images on ultrasonography, CT, PET-CT, MRI and/or ECT bone scan. The study protocol was approved by the Institutional Review Board of the participant hospital, and written informed consent for this study was obtained from all patients.
Each of these 74 samples was labelled with Cy5 and was co-hybridized with pooled normal controls onto an Agilent Human Genome CGH Microarray Kit 244A, and the copy number levels for each of the 244K probes on the array were measured. Using the raw aCGH data, we first computed the log2-ratio of background-corrected Cy5 signal to background-corrected Cy3 signal for each probe on each of the 74 arrays. A simple median normalization approach was used to adjust the log2-ratios such that their median was 0 for each array. The CBS algorithm was applied to each sample separately to identify segments having different copy number levels. The default values of the parameters in the DNAcopy package were used. The outputs are the starting and ending positions, and the smoothed log2-ratios for each identified segment. The copy number levels are expected to be normal for non-tumor samples. Therefore the gain/loss calls (

where
Results
We applied the proposed method on both simulated data and a liver cancer data set. We realize that the accuracy of defined test regions depend on which segmentation algorithm is used to locate DNA copy number changes. However the sensitivity and the specificity of different algorithms are not the focus of this paper. Here, the R package DNAcopy which implements the non-parametric CBS algorithm5,6 was used for segmentation as it has been shown to have good operational characteristics.14,24
Simulation study
We performed simulation study on 90 consecutive probes. The copy number status for probes 1 to 30 and 61 to 90 are normal for all 63 subjects. For probes 31 to 60, on average 50% of the 63 samples have copy number aberrations. Suppose the clinical event of interest is cancer metastasis. When a probe or region is associated with the clinical event, subjects have different chance to get metastasis depending on their copy number status. To be specific, the probability of having cancer metastasis is 2/3 for subjects who have CNAs, and 1/3 for subjects who have normal copy number status. The raw log2-ratios were generated from N(μ, σ 2 ), where μ = 0 for normal copy number status, μ = log2 (3/2) = 0.585 for CNAs, and σ = 0.167 was estimated from the liver cancer study discussed below. Segmentation was performed on these 63 profiles and a copy number gain was called when the smoothed log2-raio is greater than 0.5. Logistic regression was applied to each probe when the raw log2- ratios were used and to each region when the smoothed log2-ratios were used. For the gain/loss calls, Fisher's chi-square test was applied at each smoothed region. The false discovery rate (FDR) controlling procedure developed by 20 was used to control the proportion of falsely rejected probes or regions.
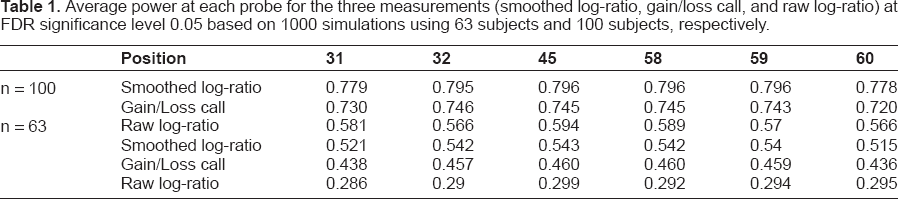
Figure 2 shows the mean sensitivity over 1000 simulated datasets for different thresholds of FDR, where sensitivity was computed as the proportion of correctly rejected probes among the total number of truly metastasis-associated probes. It is evident that both analysis, smoothed log2-ratio and discretized gain/loss calls, performed on segmented regions resulted in greatly improved power and sensitivity comparing with the analysis using log-ratios on the probe level, while smoothed log2-ratio yielded the highest sensitivity. Figure 3 shows the average power over 1000 simulated datasets for each of the 90 probes at FDR significance level 0.05. It can be seen that region-based testing methods yielded improved power than probe-based tests for every probe, although the power reduced for probes close to the breakpoints of the regions due to the variation of the segmentation methods. Table 1 shows the average power comparisons using smoothed log-ratio, gain/loss call, and raw log-ratio for probes 31 to 60 which are truly associated with the event, at FDR level 0.05 for sample sizes 63 and 100 respectively. It is clear that when the sample size increases, the statistical power increases no matter what measurement is used (smoothed log-ratio, gain/loss call, or raw log-ratio), and the performance of the gain/loss calls gets closer to that for smoothed log-ratios. However the underlying assumption for gain/loss calls being more powerful than the raw log-ratios is that the gain/loss calls are made correctly. Otherwise, using gain/loss calls may lose power or even lead to the wrong conclusions.

Average sensitivity versus different FDR thresholds based on 1000 simulations. Effects of the three types of DNA copy number measurements on the mean sensitiviy: smoothed log2-ratio (square), gain/loss call (diamond), and raw log2-ratio (circle).

Average power at each probe using FDR significance level 0.05 based on 1000 simulations using 63 subjects. The three types of DNA copy number measurements: smoothed log2-ratio (square), gain/loss call (diamond), and raw log2-ratio (circle), are compared with respect to average power at probe.
Average power at each probe for the three measurements (smoothed log-ratio, gain/loss call, and raw log-ratio) at FDR significance level 0.05 based on 1000 simulations using 63 subjects and 100 subjects, respectively.
Results for liver cancer data
Using normalized log2-ratios
In this paper, we focus on identifying CNAs which are associated with distant metastasis using three different types of measurements of copy number levels. We first performed the analysis using normalized log2-ratios.
When the FDR level was increased to 0.30, there were 2441 statistically significant probes. CNAs usually occur on a relatively large genome region, one or two probes alone being statistically significant do not give reliable results for CNAs. Number of statistically significant probes was reduced from 2441 to 695 by restricting our examination to genome regions with at least three consecutive statistically significant probes.
Using smoothed log2-ratios
The total 226,000 probes were divided into 13,566 test regions after dropping the segments on sex chromosomes. Excluding the test regions which contain one or two probes only, we performed analysis on the remaining 10,258 test regions. At FDR level 0.10, we identified 119 statistically significant test regions covering a total of 2593 probes. The number of probes covered by each of the 119 test regions varies from 3 to 114.
Using gain and loss calls
We performed log-rank test using gain/loss calls to identify chromosome regions on which patients with different copy number status have different survival curves of metastasis. Non-variable test regions where the copy number levels are normal for all 63 patients were excluded from further analysis. We also excluded the test regions containing one or two probes only. Finally, from the clinical standpoint, a log-rank test is performed on a test region if and only if there are at least 6 subjects (6/63 ≈ 10%) in at least two of the three copy number status (gain, normal, and loss). If the frequency of CNA is less than 10% for a copy number status at a test region, patients having the corresponding status will be excluded from the log-rank test on that region. This is very important as extreme values may have a huge effect on the log-rank test. It is important to note that different patients may be excluded at different test regions. To summarize, there were 4190 test regions and the log-rank tests were performed on 1409 of them, total of 10 statistically significant test regions covering 1163 probes were yielded at FDR level 0.10.
Comparisons of methods
Since using normalized log2-ratio does not yield any statistically significant regions with FDR level below 0.269, we compared results using normalized log2-ratio at FDR level 0.30 with those for smoothed log2-ratio and gain/loss calls at FDR level 0.10 (Fig. 4). For the purpose of comparisons numbers of probes covered by statistically significant test regions were listed. The three types of measurements of copy number levels gave overlapping results, and analysis using the smoothed log2-ratio yielded the largest number of statistically significant results. The identified metastasis-associated CNAs in common by all three methods are on the short arm of chromosome 12. In addition smoothed log2-ratios identified regions on chromosome 2. Statistically significant results detected by normalized log2-ratio or gain/loss calls alone consist of a relatively small proportion of probes.

Numbers of statistically significant probes for the liver cancer study. The three types of DNA copy number measurements: smoothed log2-ratio, gain/loss call, and raw log2-ratio, are compared in terms of identification of statistically significant probes.
Detection of significant CNAs in association with metastasis
The association between clinicopathological variables and metastasis in 63 patients with HCC is summarized in Table 2. Among the 7 variables evaluated, only tumor stage (
Clinicopathological characteristics and their association with metastasis.

According to the Edmondson-Steiner grading system
Cox proportional hazard regression model, adjusted for each other.
The copy number aberrations (CNAs) in association with metastasis.

Cytoband and map position are based on the public UCSC database [Human Genome Browser, May 2004 Assembly (hg 17)]
Cox proportional hazard model with the adjustment for sex and tumor stage.
Discussions and Conclusions
Chromosomal aberrations such as gains, losses, structural rearrangements and other genetic mutations are hallmarks of human cancers.3,25 Among them, genomic CNA has been regarded as an essential component in multiple types of cancers including HCC. 26 CNAs may contribute to the development and progression of various cancers by inducing gene expression alterations with or without other genetic mutations. 25 Identifying genomic CNAs in association with clinicopathological characteristics may provide some insights into the initiation and progression of cancer, and improve the diagnosis, prognosis, and treatment strategies.
In this paper we discussed the statistical framework for downstream analysis in copy number studies, using identification of CNAs associated with cancer metastasis as an example. When downstream analysis is performed at probe level and the correlation structure among adjacent probes is ignored, the power for detecting clinical event-associated CNAs is reduced. Here, we defined test regions based on DNA copy number patterns across samples, using either smoothed log2-ratios or discrete data of gain/loss calls. Segmentation could be done for each sample separately, or for all samples simultaneously. Downstream analysis such as survival analysis is performed on these test regions instead of individual probes yielding improved power due to reduced number of tests. The advantages of using test regions instead of probes are at least twofold. One is that the number of tests performed is reduced from hundreds of thousands of probes to thousands of test regions. The other is that the adjacent test regions are not correlated with each other, and the false discover rate controlling procedures can be applied without losing power.
We further compared the effects of using different types of copy number measurements on downstream analysis. The raw log-ratios are usually very noisy even after normalization. The large number of probes makes it more difficult to detect statistically significant CNAs with limited number of subjects. New multiple testing methods which can take into account the correlations among the neighbouring probes are needed. Gain/loss calls are convenient for medical practitioners to explain the clinical significance. Survival curves for different copy number status (gain, loss or normal) can be visually presented. However the results are heavily relied on which threshold is used to call a test region having copy number gain or loss. In our analysis, we used the copy number measurements from non-tumor (control) samples as the reference. In many experiments, the control samples are not included in the experiment. In addition, summarizing log2-ratios into gain/loss calls may lose information present in the original measurements. 27 When inappropriate criterion is used, it may lead to unreliable categorization of true copy number level; hence the threshold used for making gain/loss calls has a deterministic effect on downstream analysis. For a potential improvement, the uncertainty of gain/loss calls should be incorporated into downstream analysis, which would be a topic for future research. From the simulation study and real data analysis, smoothed log2-ratio has the largest power in detecting genome regions with CNAs associated with clinical outcomes due to the reduced number of test regions from the huge number of probes, and the accurate and reliable measurement of copy number levels by borrowing strength from neighbouring probes.
Array-based CGH analyses of HCC have identified a number of recurrent chromosomal CNAs and some of these have been associated with tumor stage, differentiation, and survival.26,28,29 However, no study has focused on the identification of the CNAs which play a key role in the determination of distant metastasis of HCC patients. We reported here for the first time that chromosome region loss on 3p14.2, 5q13.2, 7p15.2, and 12p13.31–11.22 are associated with distant metastasis of HCC. It has been reported 12p loss was frequently found in high-grade tumors and recurrent tumors in uterine leiomyosarcomas.
30
Furthermore, loss on 12p13.31 has been reported to be one of the most powerful independent markers for poor outcome in multiple myeloma.
31
Taken together, these results indicated that 12p loss is associated with the progression of malignant tumors, in support of the present observation of 12p13.31–11.22 loss in association with distant metastasis in HCC. In the 19.97 Mb window within 12p13.31–11.22, there locates many cancer-related genes including
In this paper, we have presented a simple but efficient dimension reduction method to identify genome regions with CNAs associated with clinical outcomes. We recommend the use of smoothed log2-ratio for downstream association test because it demonstrated the best statistical power. Even though the proposed method is demonstrated using aCGH data, the concept of test region can be applied to cancer-related CNAs studies using SNP arrays or sequencing technologies. This method has the potential to be applied for clinical screening of CNAs, thus help develop more accurate strategies in diagnosis, prognosis and therapy.
Disclosures
Author(s) have provided signed confirmations to the publisher of their compliance with all applicable legal and ethical obligations in respect to declaration of conflicts of interest, funding, authorship and contributorship, and compliance with ethical requirements in respect to treatment of human and animal test subjects. If this article contains identifiable human subject(s) author(s) were required to supply signed patient consent prior to publication. Author(s) have confirmed that the published article is unique and not under consideration nor published by any other publication and that they have consent to reproduce any copyrighted material. The peer reviewers declared no conflicts of interest.
Footnotes
Acknowledgments
ZZZ's work was supported by the Medical Science and Technology Innovation Fund of PLA, Nanjing branch, China (No. 08MA023; No. 09MA022), and Ningbo Nature Science Foundation Program (No. 2009A610126).