
Abstract
Advances in next generation sequencing (NGS) and mass spectrometry (MS) technologies have provided many new opportunities and angles for extending the scope of translational cancer research while creating tremendous challenges in data management and analysis. The resulting informatics challenge is invariably not amenable to the use of traditional computing models. Recent advances in scalable computing and associated infrastructure, particularly distributed computing for Big Data, can provide solutions for addressing these challenges. In this review, the next generation of distributed computing technologies that can address these informatics problems is described from the perspective of three key components of a computational platform, namely computing, data storage and management, and networking. A broad overview of scalable computing is provided to set the context for a detailed description of Hadoop, a technology that is being rapidly adopted for large-scale distributed computing. A proof-of-concept Hadoop cluster, set up for performance benchmarking of NGS read alignment, is described as an example of how to work with Hadoop. Finally, Hadoop is compared with a number of other current technologies for distributed computing.
Keywords
Introduction
Recent advances in high-throughput technologies, including next generation sequencing (NGS), mass spectrometry (MS), and imaging assays and scans, are providing unprecedented capabilities for cancer researchers to interrogate biological systems of interest, while creating tremendous challenges with respect to data management, access, and analysis. The Cancer Genome Atlas (TCGA) project, 1 for example, currently provides germline and tumor DNA-sequencing, RNA-sequencing, methylation, and imaging data from thousands of patients across multiple solid tumor and hematologic malignancies. Consequently, cancer researchers are faced with the formidable task of managing and integrating massive amounts of data, produced in structured as well as unstructured formats, to be positioned to use this treasure trove of data to push the scientific envelope. The requisite analyses are not confined to traditional assessment of differential expression but extend to integrative genomics including analysis of expression quantitative trait loci (eQTL 2 ) linking DNA and RNA sequencing data.
In many cases, the data volume, velocity, and variety 3 generated by these high-throughput platforms have collectively rendered the traditional single- and cluster-farm computing model, which was employed with great success in the microarray and genome-wide association studies (GWAS) era, technologically obsolete. Recent advances in computational technologies, especially distributed computing for “Big Data”, such as Hadoop, have shown great potential as technological solutions for addressing the challenges of the data deluge in next generation cancer research.
This paper provides an overview of scalable and distributed computing technologies with specific emphasis on the widely used open source Hadoop project. The presentation is organized as follows. In the next section, we provide an overview of the elements of scalable computing systems and provide a number of examples. Afterward, we provide an introduction to Hadoop as a full-featured distributed system for scalable computing and data storage and management. This section also includes an overview of the Hadoop ecosystem and specific examples of bioinformatics applications leveraging this technology. In the section that follows, we outline a proof-of-concept (POC) cluster to illustrate the design and implementation of a basic NGS data pre-processing system based on Hadoop. In the Discussion, we consider other available and widely used systems for distributed computing that could be used as an alternative to or in concert with Hadoop depending on the specific cancer informatics challenge at hand.
Scalable Computing Systems
Background
Computing models
Broadly speaking, computational systems can be grouped into two categories (see for example Refs.4,5):
Heterogeneous systems: These are typically single node workstations or servers for which computational power is scaled by upgrading or adding additional Central Processing Units (CPUs) or memory along with other components including Graphics Processing Units (GPUs) or Many-in-Core co-processors. Homogeneous distributed systems: Another way to scale computation is by connecting several computers. If the computers are connected within the same administrative domain, the collective is referred to as a compute cluster. If connected across networks and administrative domains, it is referred to as a computer grid. The individual computers in the collective are called nodes. Scaling a cluster or grid is typically accomplished by adding nodes rather than adding components to the individual nodes.
Scaling of computation is accomplished through task or data parallelization.6,7 In task parallelization, a computational task is divided into several tasks to be run in parallel on the same dataset and the results are combined. For large datasets, this approach is often not feasible as the data may not fit into memory. In data parallelization, the data are divided into smaller sets and the same processing is applied to each subset after which the results are combined.
Traditionally only a single task or instruction could be carried out on one piece of data and the related CPU architecture is called Single Instruction Single Data (SISD 8 ). In order to enable parallelization, other architectures have been developed. These include Single Instruction Multiple Data (SIMD 8 ), which allows the same instruction or processing to be applied to different datasets, or Multiple Instruction Multiple Data (MIMD 8 ) in which multiple instructions can be applied to different datasets.
In a distributed system, processing is done by the CPU in each of the nodes. Although the CPUs of the individual nodes are independent of each other the memory and storage could be shared among the nodes. The data stored on disk are made available to the CPU for processing through the memory, which requires a means of transferring the data through a communication channel such as memory bus and inter-node networking. If data are shared between the nodes, then coordination between the processes running on different nodes is required to maintain a consistent state of the data. The three common architectures for distributed systems are 9 :
Shared nothing, in which each node in the cluster works independent of other nodes with no inter-dependence for memory or storage. If required, coordination among processes running on different nodes is accomplished by passing messages among them or by underlying distributed system management software called middleware. An advantage of this configuration is that because of lack of dependence among different nodes, the cluster can scale indefinitely by simply adding more nodes to the cluster.
Shared memory, in which the nodes have access to a common memory that can be used for coordinating the processing tasks among the different nodes.
Shared disk, in which data processed by the different nodes are shared either through a central storage or by direct exchange with each other.
In distributed systems, the bandwidth of the communication channels used for sharing data and passing messages as well as the processing overhead of maintaining data in a consistent state can have a significant impact on the performance of the system. 9 Each of the three distributed system architectures employs a different strategy for managing processing. The shared-nothing architecture has the advantage of minimizing network latency and process coordination for data consistency because of the independence of the nodes for memory and data.
Data storage and models
Efficient data storage, representation, and management is crucial for building a high performance scalable system. Data storage devices can be connected to a computer in one of three different ways:
Direct attached: Storage is attached directly to the computer through an interface including Serial AT Attachment (SATA) or Serial Attached SCSI (SAS). Network attached: Storage is attached to a local network, such as a LAN, and other devices in the network can access the storage device through interfaces such as Fiber Channel. Remote: Storage is physically located outside of the internal network and is accessed through the Internet as in cloud storage. Amazon Web Service, for example, provides cloud storage through their S3 service.
10
Data required for active use are usually stored in “on-line” primary storage, which provides fast access and availability on demand. Data not requiring access for an extended period of time and that can be archived are usually stored on slower and relatively inexpensive “off-line” tertiary storage. For infrequent access, data can be stored in “near-line” secondary storage. 11 Arrangement of data storage among these tiers optimizes the storage cost without compromising ready availability of data for processing. Hierarchical storage management (HSM) software, such as IBM Tivoli, 12 automatically manages the storage and movement of data in a tiered hierarchy of devices. Data storage, access, and transfer are particularly challenging in systems that have distributed storage and clients that require data access.
A data model is a logical, rather than a physical, representation specifying the structure, types, and relationships among data elements. Applications can use data models to access data based on the logical representation without concern for the physical storage location or media. For example, in a Relational Database Management System (RDBMS), the Structured Query Language (SQL) is used for data access and manipulation based on the relational data model. 13 Specific data models, along with tools and Application Programming Interfaces (APIs) to manipulate the data, have been created for scientific computing. Two data models that have been used extensively by the scientific community are the Network Common Data Form (NetCDF) and the Hierarchical Data Format (HDF 15 ). Graph database models16,17 are used extensively for representing highly interconnected data. The Resource Description Framework (RDF 18 ) provides a set of specifications for a graph data model for use on the web. The data elements are subject-predicate-object expressions called triples or triple stores. 17 Each of the three components is a web resource represented by a Uniform Resource Identifier (URI). SPARQL 19 is the standard querying language for an RDF triple store.
Higher level information about data is provided by metadata, which can be syntactic or semantic. 20 Syntactic metadata provide information about the data structures, formats, and other physical characteristics of the data including file names and organizational hierarchy. Semantic metadata provide information about the meaning of the data elements particularly in the context of the knowledge domain. Both syntactic and semantic metadata can range from simple to complex. At a simple level, syntactic metadata are used by file systems to manage file storage and access. At a complex level, they are used by programs and databases to define data structures and schemas to be used by applications for data queries, manipulation, and analysis. Semantic metadata at a simple level provide definitions for data elements through naming classifications, data standards, and terminologies such as taxonomy, vocabulary, dictionary, and thesaurus. 20 These can be used for effective data sharing, data integration, and interoperability among applications. More sophisticated semantic metadata, including ontologies, provide complex representations of the data to include relationships to be used for knowledge inference. 21 The National Cancer Institute (NCI) has created a terminology called NCI Thesaurus (NCIt 22 ) specifically for cancer research, which contains clinical and research terms related to cancer. Concepts for drugs, therapies, and genes, among many others, that are contained in the NCIt include terms, codes, synonyms, and relationships among the concepts. There are more than 200,000 relationships among more than 55,000 concepts, which can be used, among other things, for performing integrative analyses of data from different cancer research experiments.23,24 Several other thesauri are available for clinical research including the Unified Medical Language Systems (UMLS) meta-thesaurus and the Systemized Nomenclature of Medicine Clinical Terms (SNOMED CT). 25 Gene Ontology is one of the most widely used semantic metadata in genomics research, which evolves dynamically with increase in knowledge about genes and proteins in eukaryotic cells. 26
RDBMS 13 has served as a predominant technology for data management. The hallmarks of an RDBMS are the atomicity, consistency, isolation, and durability (ACID 27 ) properties. In these systems, the data are stored in a highly structured format subject to enforcement by a relational schema. An RDBMS is considered to be an Online Transaction Processing system (OLTP 28 ). These systems are characterized by frequent reads and updates, referred to as transactions, of a relatively small number of data records. The performance of an RDBMS generally degrades as the number of data records or data fields increases. A key development to address the limitation of RDBMS with respect to scalability is the introduction of parallel databases using row based, also known as horizontal, partitioning. 29 These databases distribute mutually exclusive sets of rows among the nodes of a cluster. The SQL query is applied to each partition on each of the nodes. The results of the partitioned queries are sent back to a single node to be merged to produce the final result. The RDBMS has been adopted for managing and querying GWAS data. 30 An OLTP system is optimal for research projects that require a large number of small and simple queries, and frequent data updates. An On-line Analytical Processing (OLAP 28 ) system on the other hand is optimal for projects requiring complex analytical queries but not frequent updates. The performance of an OLTP system is measured on the basis of its ability to maintain data consistency and integrity while maximizing the number of transactions per time unit. The performance of an OLAP system is measured on the basis of the throughput of the query and the corresponding response time. 28 Data warehouses are OLAP systems developed to aggregate large amounts of data from multiple sources. Because of the heterogeneous sources of data, the incoming data are typically cleaned and transformed before they are loaded into the system. One of the key data warehouses in biomedical research is Informatics for Integrating Biology and the Bedside (i2B2)31,32 developed under the sponsorship of National Center for Biomedical Computing. The most common use of i2B2 is to repurpose data from Electronic Medical Records (EMRs) to be combined with clinical and genomics data.
Multi-core computing
Until the early 2000s, most motherboards housed a single CPU with a single core for processing. Later CPUs with multiple cores were developed to overcome the computational limitation of the single core design. The multi-core architecture has the advantage of providing higher clock rates because data do not have to travel across chip sets. 33 Computational tasks are typically scaled through manual or programmatic forking of tasks to multiple cores, or by writing multi-threaded applications using the Open Multi-Processing (OpenMP34,35) API.
High performance computing
Two widely used approaches for distributed computing over a cluster or grid of server nodes or workstations are message passing and batch queuing. These fall under the category of High Performance Computing (HPC). 36 Batch queuing systems simply distribute individual jobs as batches to the nodes. The job submission and management, and resource allocation are orchestrated by a batch submission engine such as the Simple Linux Utility for Resource Management (SLURM 37 ). The most commonly used system for message passing is the Message Passing Interface (MPI 38 ). This can be considered as a MIMD shared nothing distributed memory architecture as memory is not shared among the nodes and communication is done strictly through message passing. The batch queuing approach typically does not require specialized programming. MPI provides computational scaling at the cost of increased programming complexity. A number of bioinformatics applications developed for MPI-based distributed systems are listed in Table 1.
MPI-based applications for bioinformatics.

GPU computing
The GPU architecture is highly amenable to parallelized scientific computing.39,40 Development languages and APIs for General Purpose computing on GPU (GPGPU) include the Open Computing Language (OpenCL41,42), Compute Unified Device Architecture (CUDA 43 ), and Brook for GPU (BrookGPU 44 ). The CUDA language is intended for GPGPU on devices manufactured by NVIDIA. OpenCL is a standard adopted by several vendors including NVIDIA and AMD. A number of bioinformatics applications developed for leveraging GPUs are listed in Table 2. A typical approach for scientific computing on a GPU is to copy data from the host (CPU) memory to the memory on the device (GPU). The calculations are then carried out on the device after which the results are copied to the host. If the memory limitations of the device, relative to the size of the data, were to necessitate breaking up the data into smaller chunks, an overhead penalty is incurred because of repeated copies between the host and the device. Recent GPU card offerings provide larger amounts of memory, compared to early GPUs, rendering this technology now feasible for analysis of high-throughput data from NGS assays. For example, the NVIDIA K80 card 45 consists of two GPUs collectively housing 24 GB of memory and 4,992 streaming cores providing peak single- and double-precision floating-point performance of 2.91 and 8.74 teraflops, respectively. The AMD FirePro W9100 46 card consists of a single GPU housing 16 GB of memory and 2,816 stream processors providing peak single- and double-precision floating-point performance of 5.24 and 2.62 teraflops, respectively. The GPU technology can be scaled further by installing multiple cards on the same motherboard. 40
GPU applications for bioinformatics.

Cloud computing
The ability to conduct scalable computing requires the acquisition, installation, and ongoing management of a host of hardware and software resources. This may neither be a practically nor economically feasible proposition. Cloud computing has proven to be a powerful alternative for researchers to conduct scalable computing on a virtual computing infrastructure hosted and managed by a service provider. The infrastructure may consist of hardware resources including storage, CPU or GPU nodes, or software resources including middleware, applications and development frameworks provided to the researcher, who can provision the resources elastically depending on the research needs.
47
There is no standard definition for cloud computing. The National Institute of Standards and Technology (NIST) defines cloud computing as a model
48
with five essential characteristics. Resources can be provisioned on demand by the researcher without direct interaction with the service provider (
Several bioinformatics applications including BLAST, genome assembly and alignment have been adapted for cloud computing. Some of these are listed in Table 3. Generally, these applications require the user to provision the computing resources on the cloud, and manage the application configuration and deployment. Several cloud manager tools have been developed enabling users to easily provision resources on the cloud and deploy one or more tools and data to work as a single unit. These tools in effect provide a “turnkey” solution to a complete bioinformatics data analysis platform. Cloudman49,50 is a manager initially developed to facilitate the deployment of the Galaxy platform 51 for NGS data analysis on the cloud. It enables packaging of the data along with the analysis tools. Other cloud management software include StarCluster 52 and elasticHPC. 53
Cloud-based applications for bioinformatics.

The Bio2RDF 54 project facilitates integration across several databases by creating the datasets in a common RDF 18 format to be pushed to the cloud for access. The data from the TCGA project are available as “Linked Data”55,56 and can be queried using SPARQL. 19 The National Centers for Biomedical Ontology (NCBO) has created BioPortal 57 for review and updates of various ontologies available to biomedical researchers. One of the key advantages of cloud computing is the provision of the infrastructure to make these federated biomedical data available to the research community for integrated data analysis.
Hadoop for Scalable Computing and Data Management
Background and core architecture
Figure 1 illustrates the two core components of the Hadoop architecture as two main layers. 58 The MapReduce layer, shown above the dashed line, is responsible for computation and resource management while the Hadoop Distributed File System (HDFS) layer, shown below the dashed line, is responsible for storage and data management. Hadoop follows the Master-Slave architecture for managing computation and data in a distributed environment. The master node schedules and coordinates the computing tasks among the slave nodes. In a Hadoop system, the master node is referred to as the JobTracker while the slave nodes are referred to as TaskTrackers. The JobTracker also provides the software infrastructure for managing distributed computing such as resource scheduling and recovery from job failures. The master process for the HDFS layer is called the Name Node and it manages the metadata loaded and distributed to the slave nodes. The latter are called the Data nodes. One of the drawbacks in earlier versions of Hadoop was the tight coupling of the MapReduce engine and distributed computing services provided by the JobTracker.
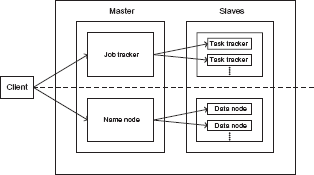
Core components of the Hadoop architecture. Adapted from Ref. 58
The computational layer of Hadoop is an implementation of the MapReduce algorithm. 59 Any implementation of this algorithm requires the provision of two user-defined functions, Map and Reduce, as its name suggests. The primary task of the Map function is to generate a set of intermediate pairs of keys and values. Before these pairs are passed on to the Reduce function, they are binned according to the keys. The Reduce function then is applied to each bin. The algorithm is often illustrated using the example of counting the occurrence of each word in a text file from the MapReduce paper. 59 Consider the example illustrated in Figure 2. The text file in this example consists of three sentences as illustrated under the Input. The file is split up along the three sentences each of which is passed on to the Map function. In this stage, the words are treated as the keys. The number of times each word appears in the sentence is the value corresponding to the key. Within each of the three bins in this stage, the keys are paired up with their corresponding value. In the next stage, Shuffle and Sort, the pairs from the previous step are grouped into bins by the keys. Each bin is passed on to the Reduce function, which adds the values from each pair therein. This effectively reduces the (key, value) pairs within each bin to a single (key, value) pair. Finally, these reduced pairs are passed on as output.
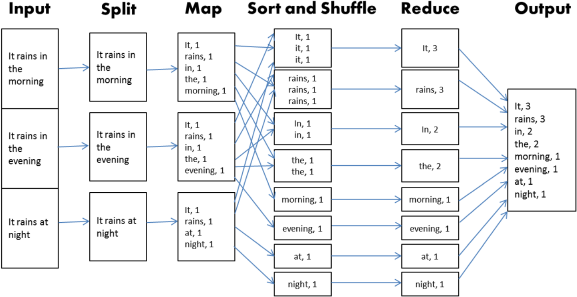
Typical MapReduce algorithm workflow.
Hadoop manages the distribution of files on individual nodes through the HDFS. Since the data are spread over a set of nodes on a network, all the complications of network programming, such as node failure, have to be taken into account. The design of HDFS facilitates the key principles of Hadoop, namely storage and management of huge amounts of data on a cluster of commodity hardware with fast access. To enable these objectives, HDFS stores very large files as blocks rather than files and provides redundancy for the data by replicating each block. The metadata for the files is stored centrally in the Name Node, which uses this information to reconstruct the files from the blocks. HDFS also works on the master-worker pattern where the Name Node is the master server and Data Nodes the workers. The Name Node, through the metadata, manages the file system namespace and the information about which Data Node has the blocks for each of the files. A Map task receives blocks of data from the Data Node and works on one block at a time. 60
Hadoop ecosystem
Decoupling of the MapReduce and the HDFS layers enables other tools to be built as higher level abstractions in other languages. Several tools have been developed to convert user applications written for these tools to MapReduce jobs for deployment and execution in a Hadoop cluster. Additionally, tools have been developed to facilitate pushing data in standard formats, including columnar data, into the HDFS layer whereby relieving the user of the burden of working with the native HDFS file format. Another major milestone in decoupling of these two layers was implemented by the introduction of the Yet Another Resource Negotiator (YARN 61 ) in 2013. 62 YARN decouples the distributed computing resource management from the MapReduce job execution engine and delegates many job flow control and scheduling functions to the individual application components. This refinement of the core architecture is expected to encourage wider adoption by allowing other computing paradigms to be implemented in Hadoop. The collection of tools built around the core infrastructure is referred to as the Hadoop ecosystem. A representative subset of tools in the ecosystem is shown in Figure 3. We provide additional details on some of the key tools next.

Representative subset of the Hadoop ecosystem.
HBase
The records in a traditional RDBMS are stored in a row format. This is generally optimal when the goal is to query a relatively small number of records (rows) consisting of large number of fields (columns). 63 For certain applications, the goal is to query a small subset of columns from a large number of records. For these, it is preferable to store the data in column format so as to increase performance of the queries. HBase 64 is a column-oriented 63 database built on top of HDFS to provide high scalability and performance in a distributed computing environment. It is modeled after Google's Big Table project. 65 HBase, unlike HDFS, provides real time read and write capability with random access for large-scale data distributed in a cluster. Linear scaling is achieved in HBase by simply adding additional nodes to the cluster. Tables in HBase can be large and sparsely populated with billions of rows and millions of columns. Columns can be added dynamically to allow for changes in the data representation. 60
Hive
Data in Hadoop are natively stored as files in HDFS. This structure does not enable convenient use of higher level languages for data queries. Apache Hive 66 offers capabilities of a data warehouse by providing the ability to represent data as tables similar to those of a relational database. 67 To this end, it provides the SQL-like language HiveQL 67 to convert queries into a series of MapReduce jobs for execution in a Hadoop cluster. Within a traditional RDBMS, data schema constraints are enforced at load time. Hive, on the other hand, employs “schema-on-read,” which checks the constraints only when data are read by virtue of a query. This approach is optimal for loading large-scale data. 60
Pig
Processing large datasets in Hadoop may require a series of data transformation steps that may be complicated to implement as Map and Reduce functions. 60 Apache Pig 68 provides higher level data structures and data transformation functions to facilitate the programming of these tasks. Pig includes the Pig Latin 69 programming language and an execution environment for running the programs. 60 The Pig execution engine converts the user written operations into MapReduce jobs at runtime. Additionally, Pig provides a set of built-in functions for a number of tasks including math and string processing. These can be modified or augmented with user-defined functions. 68
Avro
In distributed computing systems, including Hadoop, data structured as objects are transferred on the network among different nodes as streams of bytes using the process of serialization. The process of converting the byte streams back to structured data is called deserialization.60,70 These two processes are also required for writing structured data to physical storage devices. Efficiency of these processes can have an impact on the performance of a distributed application. 60 Apache Avro 71 enables efficient implementation of serialization and deserialization for Hadoop-based distributed applications by providing an API for several programming languages. Serialization frameworks require a schema for representing data structures. Avro uses the widely used JavaScript Object Notation (JSON 72 ) format 71 to provide portability across a number of languages.
Starfish
The core components of Hadoop, namely the MapReduce execution engine and the HDFS distributed storage, are extensible through their pluggable architecture. In addition, there are various procedural and declarative interfaces for interaction with Hadoop. Such an architecture provides great flexibility for extending and building distributed applications in Hadoop. At the same time, it creates an enormous challenge for performance tuning, which is required for optimal use of resources. In order for the system to adapt to changing user needs and system workloads, several system parameters require tuning. There are over 190 configuration parameters that control the running of MapReduce jobs, 73 the default values of which may not be optimal in all cases. Manually tuning the parameters for performance optimization requires expertise and can be a daunting task of trial and error. Starfish is a self-tuning system built within the architecture of Hadoop that can be used for automatic or “self” tuning. 73 It enables applications and users of Hadoop to get good performance throughout the life cycle of the data processing jobs without the need for manual performance tuning. The performance tuning is not limited to a single MapReduce job, and can be extended to workflows and workloads consisting of multiple MapReduce jobs.
Bioinformatics applications based on Hadoop
Cloudburst, an application for mapping short reads to a reference genome and released in 2009, was one of the first bioinformatics applications developed using Hadoop. 74 Since then several other applications have been developed. 75 Many of these were developed for the purpose of conducting short read alignments, while some are for further downstream processing such as RNA-seq and variant calling. In addition, some general purpose applications have been developed based on the Hadoop ecosystem that can be used for developing custom bioinformatics applications. Table 4 summarizes a number of applications useful for cancer research. Detailed information regarding five other applications is also provided.
Bioinformatics applications for Hadoop.

Hadoop-BAM
Hadoop-BAM is an application enabling access to the contents of a BAM file 76 within HDFS by virtue of providing an API as a Java library. BAM files are treated as Hadoop input and output formats. Picard 77 is a full-featured set of tools for processing NGS data. It provides extensive support for processing reads stored in the Sequence Alignment/Mapping (SAM) or BAM formats. Hadoop-BAM is built on the top of the Picard for accessing and manipulating BAM files. This feature facilitates the Picard API to be used directly within Hadoop. By distributing the processing of BAM files across a Hadoop cluster, Hadoop-BAM enables significant gains in processing time as well as linear scalability with the number of nodes in a cluster. 76
SeqPig
SeqPig 78 is an extension of the Apache Pig scripting language for development of analysis pipelines for manipulation and analysis of NGS data on Hadoop. SeqPig scripts are converted to MapReduce tasks to operate on sequencing data stored in HDFS. It leverages Hadoop-BAM 76 to provide import and export functions for common NGS data formats including FASTQ, FASTA, SAM, and BAM. In addition to input and output functionality, it offers facilities for computing base-level statistics and pile-up among other things.
Rhipe
R 79 is an open source environment for statistical computing and graphics widely used by the research community. Its powerful analytical and graphical facilities are extended by a large library of user-contributed extension packages hosted by the Comprehensive R Archive Network (CRAN 80 ). The Bioconductor project 81 provides a large collection of extension packages for genomic research. R Hadoop Integrated Programming Environment (RHIPE) is an R extension package implementing the Divide and Recombine (D&R) method on a Hadoop cluster for parallel statistical computation. 82 The D&R algorithm divides the data into subsets on which the statistical or computational procedure of interest is applied. The results from the subsets are then recombined. Hadoop is used to orchestrate these two steps while providing resource and fault tolerance management. BlueSNP 83 is an R extension package developed for GWAS on a Hadoop cluster utilizing the D&R framework provided by RHIPE. For the statistical analysis, in addition to the tests provided, users can define and implement their own tests as R functions. BlueSNP is especially useful for performing computationally intensive statistical tests for GWAS including large-scale inference using permutation resampling and eQTL analyses. Performance benchmarking of BlueSNP suggested linear scaling in proportion to the number of nodes. 83
Hydra
Mass spectroscopy based assays used in proteomics and metabolomics research generate raw data rivaling in size of those generated by NGS assays. The identification of peptide sequences from the spectra 84 is one of the major computational challenges in this field. This is accomplished by matching the spectra against known sequences in a large database. Most matching algorithms developed for this problem are amenable to parallelization. Hydra, 85 an open source Hadoop application, performs scalable peptide database search. The Each query is based on matching the mass to charge ratio score of the source spectra against those of the target peptides in the database. The latter is generated once up front and distributed to HDFS so as to be reused for all matching tasks.
Hadoop Cluster Setup
In this section, we outline a POC Hadoop cluster to illustrate the design and implementation of a basic NGS data pre-processing pipeline for RNA Sequencing data. A Hadoop cluster requires a minimum of two nodes for a distributed architecture. It can be set up on a single node in a “pseudo-distributed” mode to be primarily used for learning and testing purposes. The POC was designed using the FlexPod product line, which is an integrated commercial distributed computing system developed by Cisco and Netapp. 86 Each of the eight nodes in the cluster was a dual eight-core CPU server, providing 16 cores per node, with 128 GB RAM. One of the eight nodes was dedicated as the Name Node and another as the JobTracker. The other six nodes served as worker nodes, each functioning as a TaskTracker and a Data Node. The combined internal storage across the eight nodes was 40 TB and used for HDFS storage. A Netapp E-series storage server with 100 TB capacity was included in the POC. Since Hadoop works most efficiently with local storage rather than network storage, to avoid the latency of moving data during a MapReduce job, the latter was primarily used as a staging area for large datasets. The process of transferring data from the staging area to HDFS for processing is called ingestion. The nodes were rack mounted and connected with a Cisco Fabric Interconnect 10 Gig switch. The latter was connected to an internal 1 Gig LAN. It should be noted that since most of the communication during the execution of Hadoop jobs is among the nodes, the network throughput during job processing is not limited by the bandwidth of LAN and will take advantage of the higher bandwidth of the switch. The external storage server communicates with the cluster using the Fiber Channel over Ethernet (FCoE) protocol. Cluster management for the Flexpod line is carried out using the UCS Manager, which is cluster administrative software providing features such as addition or removal of nodes. The design of the POC is illustrated in Figure 4.
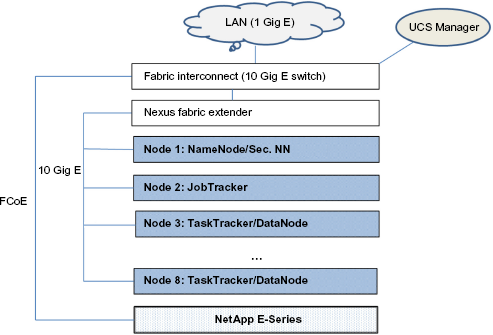
Cisco FlexPod cluster architecture.
The POC was designed to provide a scalable computing solution for a representative basic three-stage data analysis pipeline for cancer research using high-throughput sequencing assays. The primary stage is devoted to initial processing of the raw reads. These, typically stored using the FASTQ format, are aligned against a reference genome. If a reference genome is not available or a custom reference is needed, de novo genome assembly may be performed at this stage. The resulting data are captured in the SAM file 87 format. For efficient downstream processing, the file size is reduced by converting to a BAM file, a binary compressed version of the SAM file. In the second stage, the aligned reads are further processed to obtain summary information including variant calls for DNA sequencing, or gene or isoform level counts for RNA sequencing data. The third stage is devoted to downstream statistical analyses including differential expression, eQTL, and feature discovery analysis. The pipeline is conceptualized in Figure 5.
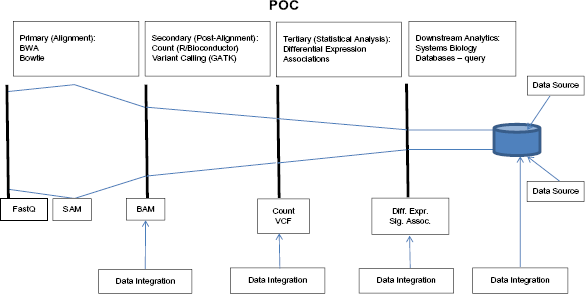
Genomics data analysis pipeline.
A preliminary performance benchmarking for the primary data analysis stage was carried out for comparing the FASTQ alignment performance on a single node with that on the POC Hadoop cluster. The single node was a Linux server with similar Alignment of FASTQ files on the Hadoop cluster done using Jnomics 88 and bowtie2 89 on the single node. FASTQ files of size 4 GB, 9 GB, and 65 GB were considered.
Discussion
To address the computational needs of a given research problem requiring a scalable computational framework, Hadoop is neither the exclusive nor necessarily always the optimal solution. Some of these are geared toward specific application areas while others are designed to provide a more generic framework. They can be used as an alternative to or in concert with Hadoop. The Genome Analysis Toolkit (GATK90,91) is a powerful and feature-rich framework for NGS variant discovery. It employs a custom MapReduce framework along with CPU threading features to facilitate variant discovery and calling. GATK uses aligned and pre-processed reads using the BAM 87 format as input and returns the results, including annotation, quality score, number of variant base calls, depth, and genotype call in the Variant Calling Format (VCF 92 ). The input BAM files can be produced using existing Hadoop applications for processing raw reads and then processed by GATK.
Genomics research in cancer often involves repeated application of computationally intensive statistical inference methods or learning algorithms on the same dataset. A performance penalty is incurred if data are written to disk after each job and reloaded from the disk into memory for the next. Spark 93 is a system for scalable computing that replaces the MapReduce layer of Hadoop while leveraging its HDFS layer. Spark enables memory resident cyclic data flow to optimize performance.94,95 The compute tasks are carried out on Resilient Distributed Datasets (RDD 96 ). An RDD is a read-only collection of objects, which can be partitioned across nodes in a cluster. The RDDs are cached in memory across the cluster nodes and can be re-used when needed in parallel without the need for writing the data to disk between individual jobs. It also provides comprehensive fault tolerance.
In case a partition is lost, it can be rebuilt from the information in the handle to the RDD about how the partition was built in the first place. The RDDs are cached in memory across the cluster nodes and can be re-used in MR jobs executed in parallel without the need for materializing the data in the RDDs to disk between individual jobs. Spark natively supports three programming languages, Scala, Python, and Java, for development and provides a library of machine learning algorithms (MLib). This library includes function for basic statistics methods, including summary statistics and random number generation, class discovery methods (eg, k-means clustering and principal components analysis
97
), and supervised learning methods (eg, support vector machines, decision tress, and random forests
98
). It also provides numerical algorithms for matrices (eg, singular value decomposition
99
) and optimization (eg, gradient descent and BGFS
100
). This library can be used as a tool kit for development of a scalable pipeline for downstream analyses. It should be noted that the HAMA project
101
provides matrix operation capabilities for Hadoop. Currently, the most widely used format for storing aligned reads from NGS assays is the SAM or its binary counterpart BAM. While there are Hadoop applications supporting this format, as previously discussed, this format may not be optimally designed for distributed computing. ADAM
102
provides a set of data formats for specifying large-scale NGS data along with a set of APIs for accessing and processing the data efficiently:
A data format and access API, built on top of Apache Avro, for transforming the general purpose genomic data formats into the ADAM format and Apache Parquet for accessing the data. A data transformation API, built using the Scala programming language and implemented on Apache Spark, for transforming and working with the data specified in the ADAM format.
Hadoop is not a turnkey technology. To gauge the performance of the POC, we considered short read sequence files of size 4, 9, and 65 GB, respectively. The maximum gain in performance was observed for the medium sized files (21 versus 50 minutes with a relative gain of 58%). The gain for the large size files was modest (227 versus 241; a relative gain of 6%), whereas for the small sized files, a degradation in performance was observed (25 versus 16 minutes; a relative loss of 56%). This degradation may be attributed to the startup overhead for operating a Hadoop cluster. It also suggests that tuning of operational parameters may be required to optimize performance for this particular size dataset. For the large size data files, the startup overhead is likely to be irrelevant. To increase performance, tuning or addition of more nodes will be required.
The computational facilities of Hadoop can be extended using other hardware and software technologies. For example, GPUs can be used in the compute nodes of a Hadoop cluster to provide a second level of parallelization. JCuda, 103 a java binding for CUDA, can be used to communicate with programs written in CUDA. 104
Conclusions
Considering the unrelenting onslaught of massive amounts of genomics, clinical, and EHR data, Hadoop provides a powerful computational framework for integrative data management and analysis in cancer research. While a number of bioinformatics tools with applications for cancer research have been developed on the basis of this technology, further refinement and development of de novo tools leveraging the collective spectrum of the Hadoop ecosystem is needed to reap its full potential. As with any other system for distributed computing, Hadoop is not a turnkey system. Developing a highly optimized solution using this technology requires understanding of computer algorithms, specifically for writing custom Map and Reduce functions, and understanding of the interaction among hardware and software components. Other technologies for scalable and distributed computing, including GPU, MPI, and Spark, are available. These can be used in concert with or as an alternative to Hadoop depending on the research problem at hand. The choice of a distributed system must be carefully evaluated during the early design stage and be periodically reevaluated throughout the course of the research.
Author Contributions
Designed the POC and carried out the pre-processing: PA. Wrote the manuscript: PA. Made critical revisions: KO. Both authors reviewed and approved the final manuscript.
Footnotes
Acknowledgment
The authors thank the reviewers for insightful and helpful comments.