
Abstract
Molecular heterogeneity within primary breast carcinomas and among axillary lymph node (LN) metastases may impact diagnosis and confound treatment. In this study, we used short tandem repeated sequences to assess genomic heterogeneity and to determine hereditary relationships among primary tumor areas and regional metastases from 30 breast cancer patients. We found that primary carcinomas were genetically heterogeneous and sampling multiple areas was necessary to adequately assess genomic variability. LN metastases appeared to originate at different time periods during disease progression from different sites of the primary tumor and the extent of genomic divergence among regional metastases was associated with a less favorable patient outcome (
Introduction
Primary breast carcinomas are known to exhibit intratumor differences in morphology 1 and expression of clinical biomarkers, such as the estrogen receptor (ER) and progesterone receptor (PR), and the extent of human epidermal growth factor receptor 2 (HER2) gene amplification.2–4 Recent studies using microarray analysis and next-generation sequencing of single cells have detected additional heterogeneity in chromosomal alterations and DNA mutations.5,6 Molecular heterogeneity within the primary carcinoma may affect diagnosis and treatment in patients where localized biopsy specimens do not accurately capture the complete genomic landscape of the primary tumor, and thus may not accurately reflect response to treatment. In addition, tumors with extensive molecular heterogeneity may be more likely to adapt to commonly used cytotoxic agents and targeted therapeutics. 7
Previous studies have detected genomic heterogeneity and discordant patterns of chromosomal alterations in axillary lymph node (LN) metastases compared to the primary breast carcinoma.8–11 Genomic sequencing revealed discordant mutations in invasive lobular carcinomas versus matched metastases 12 and enrichment of mutations in a breast metastasis from a patient with triple-negative breast cancer, 13 suggesting that cells with metastatic potential may arise from subpopulations of cells in the primary tumor. Expression of biomarkers used for defining treatment options also differs between primary carcinomas and metastases,14–16 which can pose a significant challenge to cancer medicine because treatments based on the primary tumor may be ineffective on the metastases. 17
Understanding tumor heterogeneity is critical for optimizing treatment selection and improving patient outcomes, as well as understanding mechanisms of metastatic dissemination. 18 Genetic differences between metastases and the primary carcinoma may result from early dissemination of cells with metastatic potential and ongoing molecular evolution during disease progression, or reflect the place of origin in a genetically heterogeneous primary tumor. Furthermore, evolutionary relationships between multiple axillary LN metastases remain unclear. To address these questions, we used a genomic fingerprinting approach to examine the levels and patterns of genomic changes in primary carcinomas and axillary LN metastases from 30 node-positive female patients.
Materials and Methods
Eligibility and Enrollment
Women enrolled in the Clinical Breast Care Project met the following eligibility criteria: (1) adults aged >18 years, (2) mentally competent and willing to provide informed consent, and (3) presenting with evidence of possible breast disease. Tissue and blood samples were collected with approval from the Walter Reed National Military Medical Center Human Use Committee and Institutional Review Board. All the subjects voluntarily agreed to participate and gave written informed consent. This research complied with the principles of the Declaration of Helsinki.
Tissue Collection and Characterization
All female patients meeting the following criteria were included in this study: invasive breast cancer ≥1.0 cm in diameter, multiple axillary LN metastases available for research, and no neoadjuvant treatment. Patients diagnosed prior to August 2004 (n = 11) underwent surgical excision of the primary carcinoma followed by complete axillary LN dissection. After August 2004, sentinel LN biopsy was performed, followed by axillary LN dissection (n = 19). Diagnosis of every specimen was conducted by a breast pathologist from hematoxylin and eosin stained slides; pathological characterization was performed as previously described. 19
Microdissection
Formalin-fixed, paraffin-embedded (FFPE) specimens were available from 30 patients. An average of 3.8 ± 0.8 separate pieces of primary tumor embedded in paraffin blocks and 4.9 ± 1.3 paraffin blocks containing LN metastases were used per patient. Although seven cases were included in earlier studies,8,9 all primary tumor areas and metastases were isolated and analyzed from slides created specifically for this project. The dedicated breast pathologist evaluated all the slides used for laser microdissection to identify and delineate areas within the primary tumor and metastases for dissection. Depending on cellular density, four to nine tissue sections (5 microns in thickness) were microdissected per tumor area. For each primary carcinoma, 5 to 19 areas ~2.8 mm2 in size, which were representative of the internal areas and edges of the tumor, were isolated from multiple FFPE blocks (Fig. 1A) using an ASLMD laser microdissection system (Leica Microsystems).
20
Axillary LNs were assessed for metastatic deposits by examination of an additional slide and only deposits >2.0 mm in diameter were microdissected. A single area 2.8–5.3 mm2 was obtained from each metastasis. In total, 269 primary tumor areas and 196 regional metastases were evaluated for genomic alterations.
(A) Hematoxylin and eosin stained tissue sections from a representative patient showing areas of the primary breast tumor (T) microdissected to assess genomic changes. Areas of invasive carcinoma are outlined in green. (B) Most parsimonious phylogenetic tree depicting hereditary relationships among areas of the primary breast carcinoma (T) and axillary LN metastases from the same patient shown above in panel “A” based on patterns of genomic changes assessed by Al.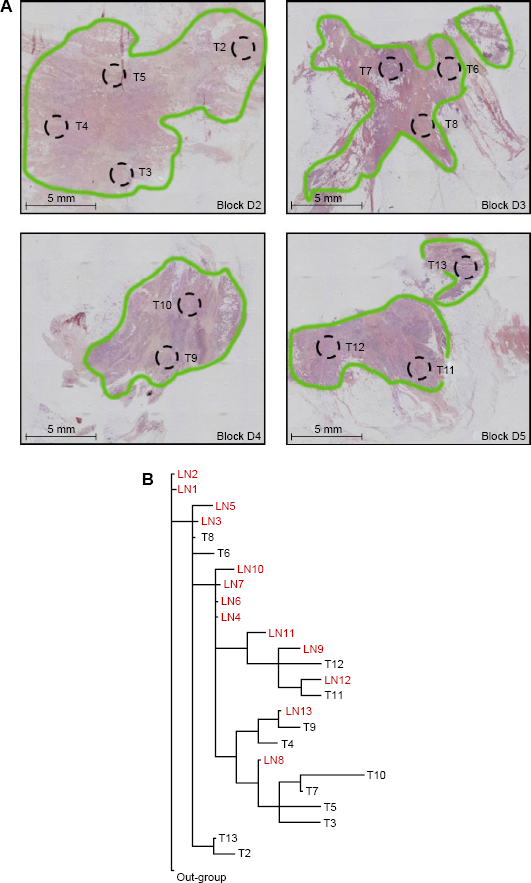
Genomic Fingerprinting
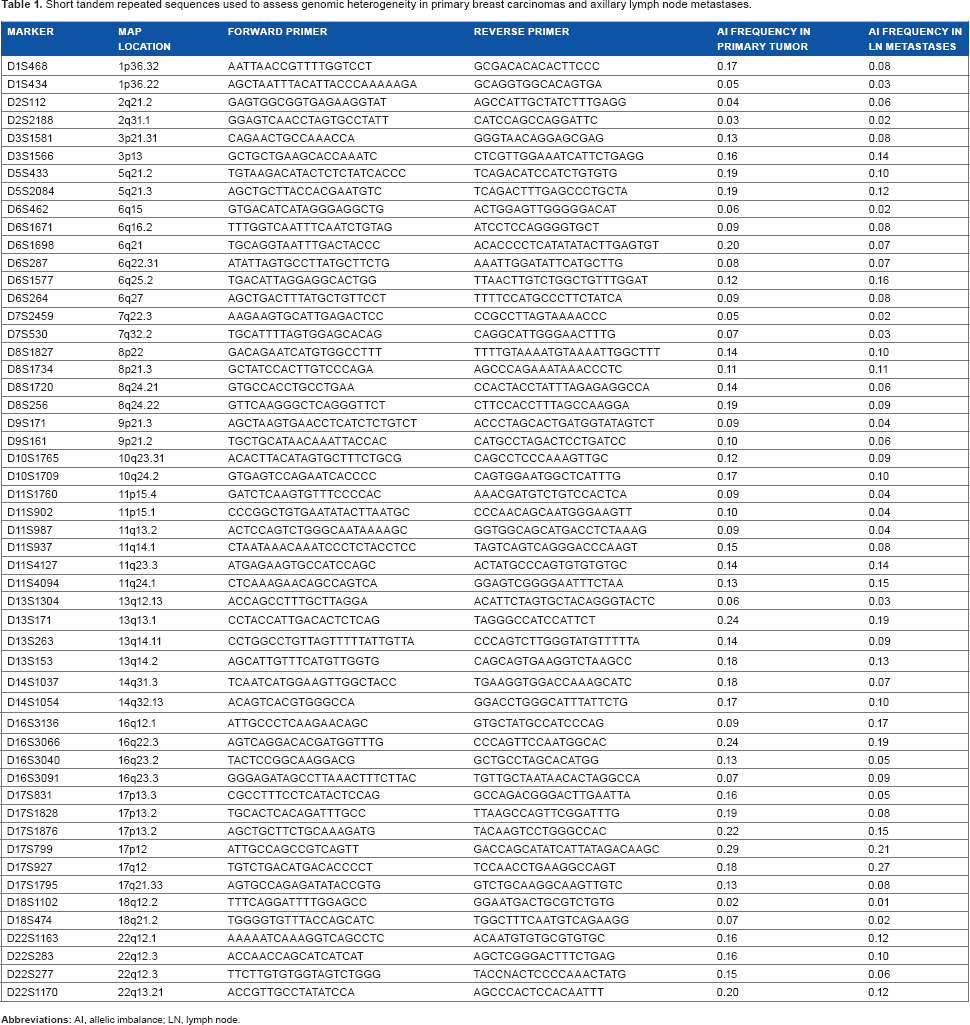
Short tandem repeated sequences used to assess genomic heterogeneity in primary breast carcinomas and axillary lymph node metastases.
Statistical Analysis
Two measures of genomic heterogeneity were calculated for all primary tumors and/or all metastases of each patient. The extent of genetic difference between tumor areas or between metastases was calculated using the mean pairwise genetic divergence score (D) as
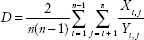
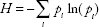
One-way ANOVA was used to evaluate relationships between genomic heterogeneity in primary tumors and axillary LN metastases and clinical/pathological characteristics with a threshold of
Phylogenetic analysis was conducted using the Phylogeny Inference Package (PHYLIP version 3.66) to determine hereditary relationships among primary tumor areas and axillary LN metastases in each patient. AI was treated as a discrete character and coded as present or absent. Analysis in PHYLIP used Wagner parsimony in the PARS program. Input options included (1) equal weighting of all chromosomal regions where AI was assessed, (2) random input order, and (3) outgroup rooting using a hypothetical (normal) tissue with no AI.
Results
Patient Characteristics and Outcomes
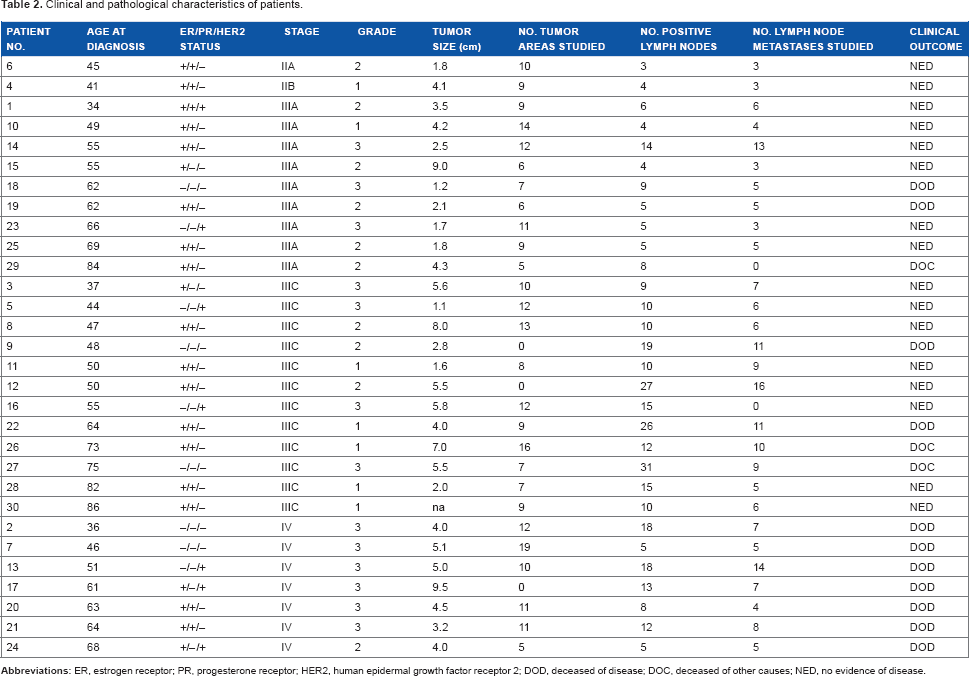
Clinical and pathological characteristics of patients.
Genomic Heterogeneity
Genomic data were available for both the primary carcinoma and the corresponding LN metastases for 25 of 30 patients; five patients had data only for the primary tumor or the axillary LN metastases. Examining a single area of the primary carcinoma captured 27% of the genomic variability (Fig. 2). Sampling at least five tumor areas was required to capture ~80% of the variability, while eight areas were needed to detect at least 90% of the genomic diversity.
Mean percentage of total AI events detectable by randomly sampling areas of primary breast carcinomas. Error bars represent 95% confidence intervals for the mean percentage of Al events detected when sampling the corresponding number of tumor areas.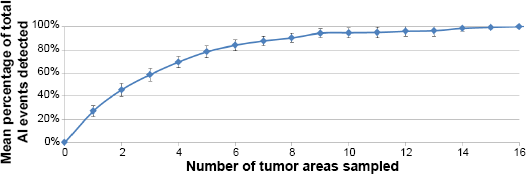
The frequency of AI events was significantly higher (
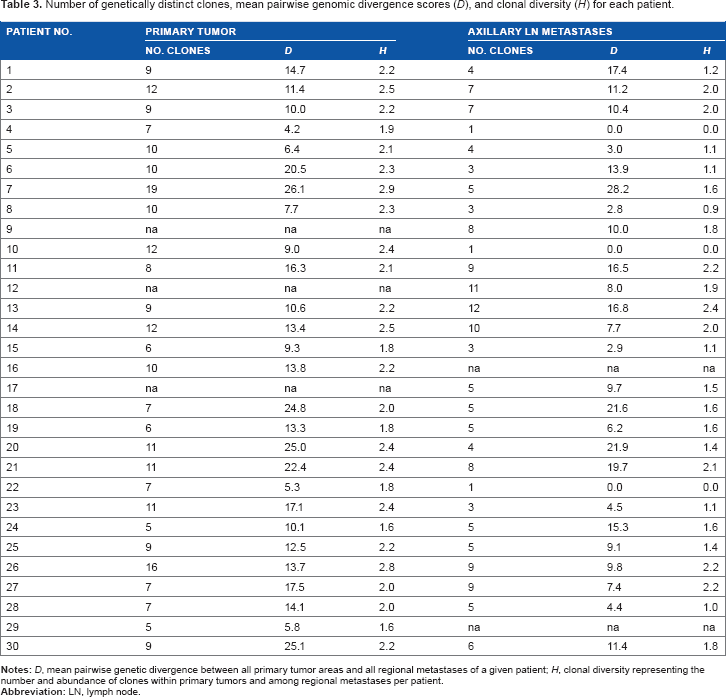
Within primary tumors and LN metastases, levels of genetic divergence were significantly correlated with clonal diversity ( (Top panel) Correlation plots of mean pairwise genetic divergence ( Number of genetically distinct clones, mean pairwise genomic divergence scores (D), and clonal diversity (H) for each patient.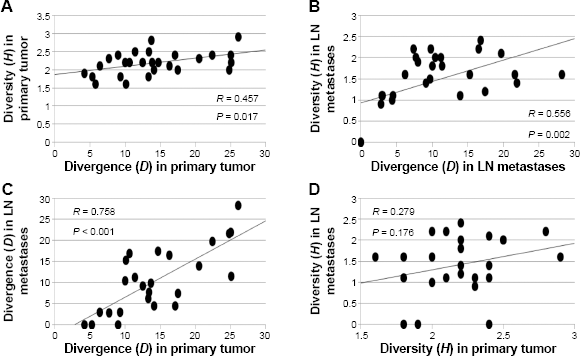
Clonal diversity (
Phylogenetic Analysis
Each primary tumor had a distinct pattern of genomic alterations, and thus a unique phylogenetic tree depicting genetic relationships among the tumor areas (Fig. 1B). Within each tumor, branch lengths were not equivalent for all areas, suggesting variability in rates of genomic change. Within each patient, LN metastases often shared a close common ancestry with specific areas of the primary tumor, indicating that the metastatic deposits in various LNs may have originated from different sites within the primary carcinoma (representative patients are shown in Fig. 4). Because these deposits carried genomic alterations already present in specific areas of the primary tumor, they likely originated later in disease progression. In contrast, LN metastases with low levels of genomic alterations did not appear to be closely related to any of the primary tumor areas sampled, suggesting that they may have been derived from cells that diverged early in disease progression before many mutations accumulated in the primary carcinoma.
Most parsimonious phylogenetic trees from three representative patients depicting hereditary relationships among areas of the primary breast carcinoma (T) and axillary LN metastases based on the patterns of genomic changes assessed by Al.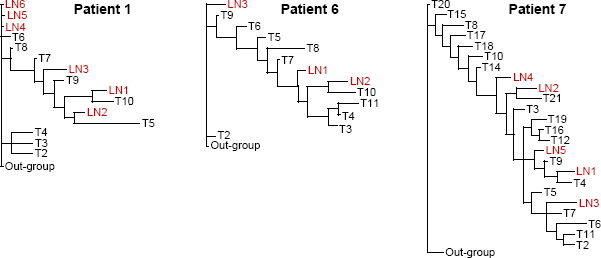
Discussion
In this study, we used a genomic fingerprinting approach to evaluate genomic heterogeneity within primary breast carcinomas and among axillary LN metastases. Multiple clonal cell lineages were observed in every primary tumor and between many metastatic deposits from the same patient. Phylogenetic analyses suggest that regional metastases likely originate independently from different sites within the primary carcinoma and may exhibit variability in the timing of metastatic dissemination.
Clonal diversification is a prominent feature of primary breast carcinomas.26,27 Heterogeneity throughout the primary tumor in patterns of point mutations and copy-number variation develops over time as clonal lineages of cells acquire genomic aberrations during tumor growth and differentiation. Tumor areas sampled here were approximately the same diameter as 14-gauge core needle biopsy specimens. The genomic diversity and divergence between these areas support the need for routine evaluation of multiple core biopsies in patient diagnosis, as pathological characterization using a single biopsy specimen would likely underestimate underlying molecular heterogeneity, which may have significant clinical consequences. 28 Genetic differences between cell lineages in the primary tumor may be responsible for the emergence of drug-resistant cells and eventual treatment failure. Because drug-resistant cells usually derive from minor subclones with different molecular characteristics than the bulk of the tumor, multiple biopsies of the primary carcinoma may be necessary to more thoroughly assess genetic heterogeneity. In this study, five tumor areas were needed to capture ~80% of the variability and eight areas were required to detect 90% of the genomic diversity.
Genetic differences between primary tumors and matched LN deposits may indicate that axillary node metastases arise from different clonal lineages within the primary tumor, and/or disseminate early in the disease process and evolve independently.8–13 Largely irreversible chromosomal changes in the primary carcinoma should be present in all cells descending from that clonal lineage. Previous studies that sampled only a single region of the primary tumor may not have accurately captured the overall clonal variability, which could affect the interpretation of genetic alterations in the primary tumor and metastatic deposits. In this study, we showed that by sampling multiple areas of the primary carcinoma, genetic ancestry of some LN metastases could be mapped to specific areas of the primary tumor, while others were not closely related to any of the sampled areas, indicating spatial and temporal variability of metastatic dissemination.
Choice of therapeutic regimen is guided by pathological evaluation of the primary carcinoma; however, metastases within a given patient have been shown to differ in response to chemotherapy and hormone therapy, 29 which may be partially explained by genomic diversity. Previous research using large-scale sequencing of a lobular breast carcinoma demonstrated that the associated metastasis contained 19 mutations not present in the primary tumor. 12 Similar observations have been reported for pancreatic cancer metastases. 30 Independent mutations may render metastases unresponsive to treatments based on the characteristics of the primary carcinoma and actionable mutations in the metastases may not be targeted. In this study, greater genetic divergence among axillary LN metastases was inversely associated with survival. This association, observed in the regional metastases but not the primary carcinoma, may be important in therapeutic resistance. Although genomic divergence among axillary metastases may be useful as a prognostic tool, less aggressive surgical approaches for managing patients with axillary disease 31 may limit accessibility to regional metastases for measuring genomic divergence. Future studies evaluating heterogeneity within sentinel LN metastases may determine whether genomic diversity is clinically useful for predicting survival.
Limitations to our study included the small number of patients with sufficient tissue from primary tumors and LN metastases available for analysis, which limited the conclusions that could be drawn from the data. Although a number of steps were taken to ensure the accuracy of AI detection, 32 DNA from FFPE samples is prone to technical artifact,33,34 and thus may not accurately estimate the number of genetic alterations. Given the small areas of tumor sampled, we examined hypervariable STR regions representing a small portion of the genome to derive molecular fingerprints of these areas and did not use global technologies such as comparative genomic hybridization or next-generation sequencing. The possibility that AI events occurred independently in separate regions and that multiple clonal lineages were present within some of the 3 mm2 regions examined may have led to an underestimation of genomic heterogeneity. Finally, these analyses were performed using tissues from advanced breast carcinomas; thus, these findings may not be applicable to patients with smaller primary tumors and/or fewer metastatic LNs.
Conclusions
Molecular heterogeneity represents a significant challenge for modern chemotherapeutics. We observed genomic heterogeneity within primary breast carcinomas and among regional LN metastases, which may have important implications for managing breast cancer patients and improving clinical outcomes. Next-generation treatment regimens should effectively target independent lineages of tumor cells that may escape current therapies in efforts to improve patient survival.
Author Contributions
Conceived and designed the experiments: REE and DLE. Conducted genomic assays: ALT, HLB, and AD. Performed pathology and histology: JAH and BD. Analyzed the data: KAM and NSC. Wrote the first draft of the manuscript: REE and DLE. Contributed to the writing of the manuscript: ALT, HLB, AD, BD, KAM, NSC, and JAH. Made critical revisions and approved the final version: REE, DLE, and CDS. All the authors reviewed and approved the final manuscript.