
Abstract
To streamline in vivo biomarker discovery, we developed a suppression subtractive DNA hybridization technique adapted for phage-displayed combinatorial libraries of 12 amino acid peptides (PhiSSH). Physical DNA subtraction is performed in a one-tube-all-reactions format by sequential addition of reagents, producing the enrichment of specific clones of one repertoire. High-complexity phage repertoires produced by in vivo selections in the multiple sclerosis rat model (experimental autoimmune encephalomyelitis, EAE) and matched healthy control rats were used to evaluate the technique. The healthy repertoire served as a physical DNA subtractor from the EAE repertoire to produce the subtraction repertoire. Full next-generation sequencing (NGS) of the three repertoires was performed to evaluate the efficiency of the subtraction technique. More than 96% of the clones common to the EAE and healthy repertoires were absent from the subtraction repertoire, increasing the probability of randomly selecting various specific peptides for EAE pathology to about 70%. Histopathology experiments were performed to confirm the quality of the subtraction repertoire clones, producing distinct labeling of the blood–brain barrier (BBB) affected by inflammation among healthy nervous tissue or the preferential binding to IL1-challenged vs. resting human BBB model. Combining PhiSSH with NGS will be useful for controlled in vivo screening of small peptide combinatorial libraries to discover biomarkers of specific molecular alterations interspersed within healthy tissues.
Keywords
Introduction
Phage display, first described by Smith, 1 has been used to identify peptides with high affinity and specificity interactions toward a wide spectrum of targets, both in vitro and in vivo. 2 Recombinant bacteriophages, which harbor combinatorial libraries of DNA with complexities at 109 order, produce the equivalent repertoire of fusion proteins displayed on their surface. Such libraries have been used to isolate organ-specific homing peptides3,4 and to discover their interacting targets.5,6 In vivo screening that targets specific pathological conditions (eg, tumor cells) produces highly complex repertoires of peptides. Next-generation sequencing (NGS) technologies have made possible the overview of their complexity7,8 but do not give access to phages expressing the corresponding markers.
We aimed to streamline in vivo biomarker discovery, using phage display, to produce the largest possible repertoire of peptides, specifically homing to tissue alterations in the central nervous system (CNS) caused by neuroinflammation, in the experimental autoimmune encephalomyelitis (EAE) rat model of multiple sclerosis (MS).
In EAE and MS, pathological tissue alterations are disseminated within the CNS and are highly heterogeneous.9,10 These characteristics make the recovery of clones of interest from a particular site impossible. Our aim was to isolate a large repertoire of EAE-specific peptides, taking into account that the EAE repertoire is recovered from the entire CNS tissue and thus contains a large number of peptides homing to the interspersed healthy tissues. To eliminate this source of noise, we developed a two-step strategy. We first performed a parallel selection of phages in healthy animals and EAE-affected animals and then used the resulting repertoires to subtract peptides homing to healthy tissues from the EAE repertoires. The physical subtraction is performed by a suppressive subtraction hybridization (PhiSSH), using the DNA of recombinant phages from the EAE and healthy repertoires. Its product, genomic DNA of recombinant phages from the EAE sample, is then used to generate a subset of the EAE repertoire, the subtraction repertoire. PhiSSH has two complementary effects: the decrease in the frequencies of highly abundant clones of the EAE repertoire and the increase in the frequencies of those of intermediate abundance, producing a subtraction repertoire that is adequate for the efficient recovery, by random picking, of a set of peptides targeting the pathological sites.
Materials and Methods
EAE immunization
Animal handling and experimentation conformed to the guidelines of the European Union (permission Nos. 6305 and 33/00055 of local Animal Experimentation Commission). Young female Lewis rats (Charles River Laboratories) were housed in cages, five animals per cage, with standard conditions of light and free access to water and food. Acute EAE was induced in female Lewis rats aged 6–7 weeks weighing around 150–170 g. Rats were anesthetized with isoflurane for 1.5–2 L/minute, and EAE was induced by intradermal inoculation of an emulsion containing 1-mg Mycobacterium tuberculosis H37Ra strain (Difco), 50-μL Complete Freund's adjuvant, and 100-μg myelin basic protein fragment from guinea pig. 11 All animals were weighed and examined daily, with grading of clinical neurological handicap symptoms according to the following scale: 0, healthy animal, no clinical signs; 1, flaccid tail; 2, flaccid tail and hind limb weakness; 3, complete paralysis of one hind limb; 4, paraplegia; and 5, death. Clinical EAE onset was defined when the rat developed a flaccid tail (score 1).
Phage peptide library and bacteria
In our study, we used a commercially available phage peptide library kit with complexity of 2.7 × 109 transformants (New England Biolabs, NEB Catalog #E8110S) with foreign random peptides of 12 aa displayed as a fusion with a minor coat protein (pIII) of M13 phage and Escherichia coli ER2738 host strain, tetracyclineresistant, F′ pilus deploying, for filamentous phage infection.
Comparative in vivo phage-displayed peptide selection from EAE vs. healthy rats
In vivo phage selection
EAE-induced rats presenting severe neurological motor dysfunction (scores 3 or 4) and healthy littermates were deeply anesthetized by intraperitoneal injection of pentobarbital (6 mg/100 g weight). Absence of spontaneous body or vibrissae movement, absence of nociceptive reflex (no movement upon hind limb pinching), and absence of behavioral reflex (no eye blinking) were ensured throughout the experiment. High-score EAE disease development was preferred, because inflammation within the central nervous tissue is most widespread in animals with more developed disease, increasing the chance of obtaining pathology-specific peptide binders compared to healthy animals, with lesions at different degrees of maturation. To generate both the pathological EAE and the healthy control phage peptide repertoires, 1 × 1010 plaque forming units (pfu) of phages of the library suspended in 400-μL 0.9% NaCl solution were administered into the blood circulation of animals. The first half of the phage solution was injected into the tail vein, and 10 minutes later, the second half of the phage solution was injected into the left heart ventricle. Two minutes after the second injection, the animals were extensively perfused with 200-mL phosphate-buffered saline (PBS) for 15 minutes to wash out unbound phages from the blood vessels.
Phage recovery from CNS
After the perfusion, the rats were dissected to extract the brain and spinal cord. Tissues were homogenized in the presence of 1% NP40 (1 mL/g tissue) in 0.9% NaCl to isolate the phages. The tissue suspension was incubated with 10 mL of E. coli bacteria ER2738 cultured in log phase (0.5 OD600) in LB broth (Sigma-Aldrich) at 37 °C for 45 minutes under vigorous shaking. Infected bacteria were further grown for four hours in 40-mL LB broth allowing phage amplification. The culture was cleared of bacteria by centrifugation (8,000 rpm, 10 minutes) at 4 °C. Phage particles from the supernatant were PEG8000/NaCl precipitated (3.33% w/v/0.42 M) overnight at 4 °C. Phages were collected by centrifugation (14,000 rpm, 10 minutes) at 4 °C and recovered in 1-mL TBS. Remaining bacteria were eliminated by a second centrifugation (8,000 rpm, 10 minutes), and phage precipitation was repeated (one hour at 4 °C). After centrifugation, phage pellets were dissolved in TBS/0.02% NaN3. Aliquots were stored at 4 °C until use.
After a first round of selection, harvested phages from each group of animals were pooled. Phage screening was then performed during three rounds of in vivo selection by injection of 1010 pfu of the previously harvested phages. Administration and amplification of the phages were performed as described earlier. All the steps of phage collection and amplification to obtain the repertoires were controlled by titration (pfu).
Subtractive hybridization for enrichment of EAE-specific phage clones
Subtractive DNA hybridization was performed to increase the concentration of phage clones expressing peptides specific to EAE tissues. Figure 1 shows the schematic representation of the physical DNA subtraction technique, involving DNA extraction from each repertoire, polymerase chain reaction (PCR) amplification of the recombinant DNA region coding for the peptides of the driver (control repertoire), and subtractive DNA hybridization (PhiSSH); the PhiSSH products were used for E. coli (ER2738) transformation to produce the subtraction repertoire.

DNA subtraction of selected phage repertoires. Single-stranded circular genomes from the EAE phage repertoire (tester) were hybridized with asymmetric PCR products from the healthy phage repertoire (driver), in conditions favoring the stabilization of hybrids with homologous peptide encoding sequences. The products of this reaction were further processed by restriction enzyme digestion with KpnI digestion, symbolized by double arrowhead, to linearize the circular molecules containing sequences common to both the repertoires. Remaining circular molecules with sequences present in the EAE repertoire are preserved and can be rescued and are propagated by E. coli transformation and culture to produce a repertoire enriched in EAE-specific sequences.
Repertoire DNA extraction
Phages were cultured after the infection of E. coli (strain ER2738, moi ~0,1) at 37 °C, with agitation (250 rpm) for 4.5 hours. Bacteria were discarded after centrifugation at 2,500 g for 10 minutes, and the phages were extracted and purified using the standard PEG/NaCl protocol. Genomic DNA was obtained by NaI lysis according to the NEB protocol (NEB, #E8110S). DNA concentration and purity were determined by spectrophotometry.
Recombinant region amplification
The recombinant region of the gIII, containing the sequences encoding for the displayed peptides, was amplified by PCR using the +18/–96 couple of primers (+18: 5′-TCGCAATTCCTTTAGTG-GTA-3′, −96: 5′-CCCTCATAGTTAGCGTAACG-3′). For the production of the subtractor DNA, a nested asymmetric PCR was performed using the +18 primer (0.1 μM) and a tenfold excess of the −28 primer (5′-GTATGGGATTTT-GCTAAACAAC-3′; 1 μM) to favor the production of single-stranded unpaired products of the negative strand; cycles were 95 °C, 30 seconds; 55 °C, 30 seconds; and 72 °C, 30 seconds for 50 cycles.
Subtractive hybridization
Single-stranded circular genomic DNA (5.3 × 10–6 M) extracted from the EAE repertoire (tester) was mixed with PCR products of the healthy repertoire (driver) in a 1:10 molar ratio in 100 μL of KpnI reaction buffer (25-mM Tris-Acetate pH 7.5 at 37 °C, 100-mM potassium acetate, 10-mM magnesium acetate, 1-mM dithiothreitol). Double-stranded (ds) DNA species were melted by incubation at 95 °C for 10 minutes, and then the temperature was slowly decreased (1 °C/minute) to reach 85 °C. Hybridization was carried out at this temperature for two hours, and then the reaction mix was rapidly cooled to 37 °C. KpnI (~800 U; Thermo Scientific) was added, and the mix was incubated at 37 °C for 10 minutes. The completion of KpnI digestion was controlled by agarose gel electrophoresis. The final products were used for bacterial transformation without purification.
Bacterial transformation and recovery of the subtracted repertoire
Chemo-competent bacteria were prepared using the RapidTransit™ Kit (Sigma-Aldrich, R2653) according to the kit's protocol. Briefly, log growth phase bacteria (ER2738) were recovered by centrifugation, suspended in 1 × RapidTransit transformation buffer, and incubated for 10 minutes on ice; then, the subtraction products were added, and the mix was further incubated for 10 minutes on ice. A heat shock at 42 °C for 30 seconds was included according to the protocol proposed by Sigma-Aldrich. Recovery medium was added, and the bacterial culture was incubated for one hour at 37 °C under shaking. Then, LB medium was added and the culture continued for an additional 3.5 hours. Bacteria were eliminated by centrifugation at 2,500 g for 10 minutes, and the supernatant containing the viral particles was saved for further processing. Transformation efficiency was of ~10e + 7 per μg of M13KE genomic DNA.
Sequencing of repertoires
Genomic DNA of the recombinant phages was extracted by the NaI method, as described earlier, from the three repertoires: EAE, healthy, and subtraction. The recombinant region of the gIII, containing the sequences encoding for the displayed peptides, was amplified by PCR using the +18/–28 couple of primers. The complexity of the resulting products was first controlled by pool sequencing (BigDye Terminator v3.1 Cycle Sequencing Kit and ABI 3130 XL; Invitrogen) and then sequenced on an Illumina Genome Analyzer IIx. All the sequencings were performed by the team of the university sequencing facility, Plateforme de Génomique Fonctionnelle de Bordeaux. Sequences were retrieved as fastq files, demultiplexed per sample, and further processed using a homemade workflow on the Galaxy platform.12,13
Processing included elimination of sequences with Phred quality scores lower than 30, reorientation of the reads to represent the positive strand of the phage, extraction of the sequences encoding for the displayed peptides, counting the occurrences, and calculation of the frequencies. For this, the DNA sequences were translated into their equivalent of peptides using the genetic code of the E. coli ER2837 strain. The peptide sequences of the three individual repertoires were quantitatively and qualitatively analyzed, and the repertoires were compared with each other.
Characterization of individual phage peptides
Phage clone selection
The EAE, healthy, and subtraction samples of the phage repertoires were grown on plates with E. coli ER2738, LB-Agar, and IPTG:Xgal. From each repertoire, about 30 clones were randomly picked and amplified in E. coli ER2738 bacteria according to the NEB protocol. DNA inserts encoding the peptides were amplified by PCR, and the products were sequenced. For both the EAE and healthy repertoires, we obtained mainly copies of the same few clones. To increase the number of phages expressing individual peptide sequences specific for EAE, we randomly picked additional clones from the subtraction repertoire. Clones expressing individual peptide sequences were chosen for further binding studies.
Phage binding on CNS sections
EAE rats developing clinical scores of 3 or 4 and healthy control rats were anesthetized with an intraperitoneal injection of pentobarbital. Animals were perfused with 50-mL PBS into the left heart ventricle, followed by 200 mL of 4% paraformaldehyde (PFA) in PBS. Brains and spinal cords were removed and maintained in 4% PFA in PBS for four days. The tissues were cut in slices of 30 μm by a vibratome (Leica VT1000S) and kept in PBS/Na azide 0.02% at 4 °C until use.
Brain and spinal cord slices from EAE and healthy rats were permeabilized with Triton 0.3% and saturated with 3% goat serum in PBS for one hour. Then, the tissues were incubated with clonal preparations (1010 pfu) of phages for two hours at room temperature (RT) under shaking. The buffer containing unbound phages was removed and the tissues were washed three times for 10 minutes with PBS. Tissues were incubated for double labeling with 12 μg/mL rabbit polyclonal to M13 bacteriophage (M13pVIII; Abcam ab6188) and 2 μg/mL mouse monoclonal antibody RECA-1 (Abcam ab9774) to endothelial cells for two hours at RT. Tissues were washed three times for 10 minutes and thereafter incubated with 3 μg/mL goat secondary polyclonal antibody to rabbit IgG (labeled by Cy3®; Abcam ab6939) and 6 μg/mL goat anti-Mouse IgG (labeled by Alexa Fluor® 488; (Abcam ab150113) for two hours in the dark at RT. After 3 × 10 minutes of washes in PBS, tissues were incubated with 10 μg/mL DAPI (Euromedex ref 1050) in PBS for 10 minutes and washed two more times for 10 minutes with Tris-HCl 0.05 M pH 7.5. Finally, sections were mounted on glass slides with VECTASHIELD® Mounting Medium (CliniSciences). The sections were viewed by epifluorescence microscopy (90i Nikon) and, for details, by confocal microscopy (SP8 Leica).
Phage binding on hCMEC/D3 cells
Human endothelial cell line hCMEC/D3 (kindly provided by Dr. Pierre-Olivier Couraud, Institut Cochin, Paris) was grown as a confluent monolayer on rat tail collagen (Life Technologies) on precoated LABTEK chamber slides (Dutscher), following the established protocol at 37 °C, 5% CO2 in EBM-2 basal medium (Lonza) containing 5% decomplemented fetal calf serum (PAA), 10-mM HEPES (Dutscher), 1 ng/mL of basic fibroblast growth factor (Sigma-Aldrich), hydrocortisone (1.4 μM), ascorbic acid (5 μg/mL), chemically defined lipid concentrate (CONC, Life Technologies), and 1% antibiotics (penicillin-streptomycin), by replacing the medium every two to three days. 14
To simulate inflammation conditions, hCMEC/D3 cells were stimulated with recombinant human IL1-β (rhIL1-β). Various stimulation time points (2, 4, 8, 16, and 24 hours) with various rhIL1-β concentrations (5, 20, and 100 ng/mL) were tested in a procedure similar to that described by Hurst et al. 15 Stimulated cells were then incubated with three phage clones from the subtraction repertoire and the wild-type (wt) phage as control; we observed differences in binding starting already at two hours and increasing with time exposure. Most variable binding was observed at four and eight hours. For our experiments of hCMEC/D3 stimulation, we chose five-hour exposure with 20 ng/mL rhIL1-β. Controls with hCMEC/D3 in resting state were not stimulated. Single-phage clones (1010 pfu) were incubated with cells. Unbound phages were cleared by washing with PBS (three times, 10 minutes). Cells were then fixed (PFA 4%, 30 minutes) and washed twice with PBS. Cells were permeabilized with Triton 0.3% and saturated with 3% goat serum in PBS for one hour.
Thereafter, the cells were incubated with 12 μg/mL rabbit polyclonal antibody to M13pVIII (Abcam) for two hours at RT. Cells were washed three times for five minutes with PBS and then incubated with 3 μg/mL goat antirabbit polyclonal secondary antibody labeled by Cy3® (Abcam; two hours in darkness at RT) and washed again for three times.
Cells were incubated with 10 ng/mL DAPI for 10 minutes and washed twice for 5 minutes with Tris-HCl 0.05 M pH 7.5. Finally, after the removal of LABTEK chamber slides, cells were mounted with VECTASHIELD® Mounting Medium and the cover glass was fixed with nail varnish. The cells were viewed by fluorescence microscopy (Nikon 90i) at 20 × magnification. Binding studies of phage clones with hCMEC/D3 cells were repeated at least three times.
Phage dot-blot binding on hCMEC/D3 protein extracts
Confluent hCMEC/D3 endothelial cells were stimulated with or without 20 ng/mL rhIL1-β for five hours. After three washes (five minutes with PBS), cells were lysed by scraping in 1-mL nondenaturing lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 5 mM EDTA containing a cocktail of mammalian protease inhibitors; Sigma-Aldrich) to extract proteins on ice. After lysis, a short centrifugation eliminated possible remaining debris, and protein content was quantified (protein assay kit, Thermo Scientific). Equal volumes (1 mL) containing equal amounts of protein extracts (0.9 mg/mL) of hCMEC/D3 cells obtained from rhIL1-β stimulation or control cell cultures in resting state were homogeneously absorbed on nitrocellulose membranes (12 × 8 cm2) Hybond C (Amersham) for 30 minutes under shaking. The membranes were briefly rinsed with TBS and allowed to dry. Nonspecific sites on the nitrocellulose membranes were blocked by soaking in 3% BSA/TBS (30 minutes at RT). Membranes were mounted in a 96-well Hybri-Dot-Manifold apparatus for parallel testing of hCEMC/D3 protein extracts with individual phage clones (1010 pfu) expressing various peptides and the control wt phage (30 minutes at RT), in three independent experiments.
After intensive washes (five times for five minutes with PBS), the membranes were incubated with rabbit antibody directed against phage M13pVIII (1:300 in PBS) for two hours. After five washes with PBS, they were incubated with goat antirabbit horseradish peroxidase-labeled antibody (1:1,000; Sigma-Aldrich A0545) in TBS-BSA (30 minutes at RT). After intensive washing, the binding of phages to protein extracts on the dot-blot membranes was revealed by 4-chloro-1-naphthol (Sigma-Aldrich) in TBS in the presence of 0.3% H2O2. The reaction was stopped by dilution with water. The detection of chromogen precipitates was measured by densitometry from the digital image of the membrane.
Results
In vivo generation of phage-displayed peptide repertoires
In vivo screening of a phage-displayed combinatorial library of 12 aa in both EAE pathological CNS and healthy CNS generated two highly complex peptide repertoires. EAE target lesions are disseminated among nonaffected tissue within the CNS. Thus, the EAE repertoire contains clones binding to both the pathological and unaffected tissues, while the healthy repertoire served as control and DNA subtractor of the EAE repertoire for PhiSSH, allowing the generation of a third subtraction repertoire. Figure 1 shows the schematic representation of the technique that is based on the single-stranded circular nature of the inoviral genome. Circular DNA-transformed E. coli is able to process the viral genome to produce the RF II necessary for phage propagation, while linear DNA molecules are rapidly degraded by bacterial exonucleases. Single-stranded DNA is not a suitable substrate for most restriction nucleases as they fail to bind. By hybridization, local ds structures can be produced that are susceptible to digestion by restriction endonucleases, including the KpnI binding site, proximal to the 5′ position of the recombinant DNA.
In silico analysis of phage-displayed peptide repertoires
PCR products containing the encoding sequence for the recombinant phage protein were sequenced. For the three repertoires, such as EAE, healthy, and subtraction, we obtained a total of some 9.65 million reads. DNA sequences and the derived peptides of the three individual repertoires were quantitatively and qualitatively analyzed, and the repertoires were compared with each other.
For the EAE, healthy, and subtraction repertoires, we recovered about 4.97 million, 3.17 million, and 1.51 million reads, respectively, with Phred scores higher than 30. The encoded peptide sequences from the three repertoires, their occurrences, and frequencies within each of the three repertoires are presented in Supplementary Table 1. The quantitative distributions of the occurrences and frequencies of the encoded peptides in the three repertoires are shown in Figure 2.
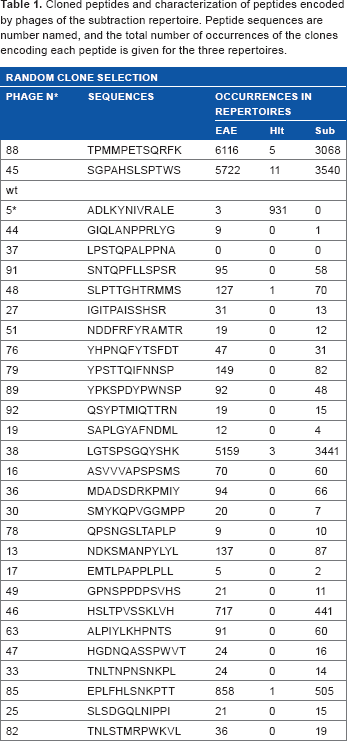
Cloned peptides and characterization of peptides encoded by phages of the subtraction repertoire. Peptide sequences are number named, and the total number of occurrences of the clones encoding each peptide is given for the three repertoires.
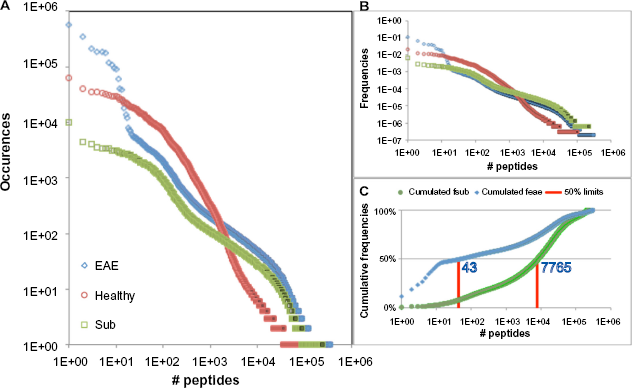
Distribution of peptides in the three repertoires, EAE, healthy, and subtraction: (
The efficiency of the repeated selection rounds of the EAE repertoire is illustrated by the presence of a small number of peptides of high abundance, covering about 47.7% of the repertoire. The EAE and healthy repertoire distributions present a crossing point at about 200 occurrences, with a large number of peptides less present in the healthy than in the EAE repertoire (Fig. 2A). Despite this dissymmetry, the efficiency of the subtraction does not seem to be affected, as we can observe by comparing the frequencies of these low-abundance clones (Fig. 2B). In fact, the parallelism of distributions of the EAE and subtraction peptides spans between the frequencies of 1e −3 and 6e −6, across the intersection point situated at ~6.5e −5, with about a twofold enrichment. The combined reduction in the frequencies of the EAE highly abundant peptides and the twofold enrichment for the large majority of the other peptides increase the probability of obtaining a large subrepertoire of EAE-specific peptides by random picking from the subtraction repertoire. Figure 2C illustrates the expected success of producing such a subrepertoire: for each random selection of a clone, there is a 50% chance that it will be 1 of the 43 more abundant ones in the EAE repertoire or 1 of the 7,765 more abundant ones in the subtraction repertoire, an increase of about 180 times in random picking efficiency.
The PhiSSH procedure was developed to enrich the presence of peptide sequences in the subtraction repertoire that are specific to the EAE pathology. The Venn diagram shows the attribution of the unique encoded peptide sequences to the EAE, healthy, and subtraction repertoires and their intersections (Fig. 3).

Venn diagram presenting the quantitative study of individual encoded peptides in the three phage repertoires: (a) EAE 304,661 peptides; (b) healthy 100,982 peptides; (c) subtraction 216,279 peptides; (ab) 5,307 common peptides in EAE and healthy repertoires; (abc) 3,613 peptides common to all three repertoires, EAE, healthy, and subtraction; (bc) 4,071 common peptides in healthy and subtraction repertoires; and (ac) 113,187 common peptides in EAE and subtraction repertoires.
Based on the evaluation of common peptide sequences within both the EAE and healthy repertoires and their presence in the subtraction repertoire, we calculated the efficacy in eliminating such common clones by PhiSSH. Among the 113,187 peptides in the subtraction repertoire that were also present in either the EAE or healthy repertoires, 4,071 peptide sequences, being also present in the healthy repertoire, remained in the subtraction repertoire, indicating the limits of the subtractive elimination by PhiSSH. These remaining (noise) peptide sequences in the subtraction repertoire represent ~3.6% of the peptide sequences that were not eliminated by PhiSSH, indicating a subtraction efficacy of eliminating common encoded peptide sequences of over 96%. Such remaining noise peptide sequences can be expected to persist for the C0t achieved during the two-hour subtractive hybridization.
To define quantitatively and qualitatively the enrichment of EAE-specific peptides in the subtraction repertoire, we considered only those peptide sequences that were identified as being present in the EAE repertoire, but absent in the healthy repertoire. Based on these data, we calculated the probability to randomly select a specific clone for EAE pathology, which was 0.39 for the EAE repertoire and 0.70 for the subtraction repertoire. We further calculated the occurrences of unique peptide sequences that are specific for EAE pathology in both the EAE and subtraction repertoires. Comparison of these EAE-specific peptides in both repertoires revealed that the PhiSSH procedure does not cause a change of their relative occurrences (Fig. 4) and thus of their frequencies (Fig. 4, insert). We further observed that the occurrences of EAE-specific clones were not higher than 1,500 in the subtraction repertoire and less than 2,500 in the EAE repertoire.

Occurrences and frequencies (insert) of EAE-specific peptides in EAE and subtraction repertoires. EAE-specific peptides of the EAE repertoire, defined as being absent from the healthy repertoire, were plotted against the same peptides being present in the subtraction repertoire. Of note, three peptides presenting major occurrences and frequencies in the EAE repertoire had only minor occurrences and frequencies in the subtraction repertoire and can therefore be considered as lost during PhiSSH.
Biotesting of phage clones
Random picking of 200 clones from the subtraction repertoire produced a list of 37 peptides, of which 28 peptides were used for biotesting, along with a clone (clone 5) specific to the healthy repertoire and the nonrecombinant (wt) phage as a control. As the copy numbers of these peptides in the three repertoires show (Table 1), most of them can be considered as EAE-specific binders, being absent or presenting very low copy numbers in the healthy repertoire.
Histopathological binding studies of selected phage-displayed peptides
To ensure that the phages were able to specifically label EAE tissues, experiments were first performed on pathological and healthy CNS sections. The phages, recombinant clones, and the wt phage (nonrecombinant), as control, were studied by immunohistochemistry with the spinal cord and brain tissue sections of healthy (Fig. 5A) and EAE-developing (Figs. 5B-5E) rats, by epifluorescence and confocal microscopy.
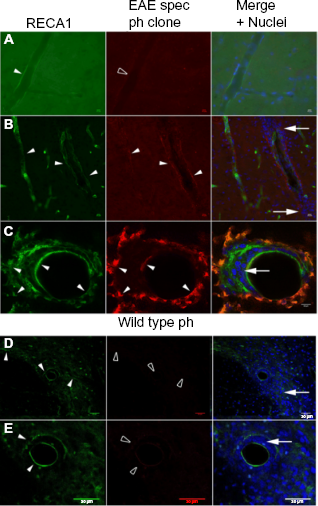
Binding of selected phage clone with EAE and healthy rat CNS. Clonal phage preparations, expressing EAE-specific peptide (EAE spec ph clone) or control wt phage (wt ph), were incubated with spinal cord sections of either healthy control rats (
No binding of these EAE-specific clones was evidenced on the CNS sections of healthy animals (Fig. 5A). Essentially, the EAE-labeling phages (example clone 88, Fig. 5) efficiently and specifically bind to blood vessels in the spinal cord and brain tissue of EAE-developing rats, which was confirmed by double labeling with RECA-1, a rat pan-endothelial cell marker. In particular, the labeled vessels present parenchymal infiltration by immune cells, which is a characteristic for EAE neuroinflammation. The case of clone 88, shown in Figure 5B and 5C, presents an even distribution of its target(s) to the luminal and abluminal locations. As expected, no labeling was observed with the wt phage on EAE CNS tissue sections (Figs. 5D and 5E).
Live binding of selected EAE-specific phage clones to human BBB in vitro model
Phages binding to blood vessels in the CNS of EAE rats indicate potential inflammationinduced molecular alterations of the BBB. To identify those clones effectively targeting altered endothelial cells, we further studied them using the hCMEC/D3 cell line, an in vitro model of the BBB [14]. Confluent cells were stimulated with rhIL1-β simulating proinflammation conditions or were maintained in a resting state. Cell cultures were then incubated with the selected phage clones, and the bound phages were revealed by indirect immunofluorescence. Figure 6 shows the in vitro binding studies to endothelial cells hCMEC/D3, with a representative EAE-specific clone and the wt phage, as control. The EAE-specific phage has low binding capacity to the endothelial cell line in the resting control state (Fig. 6A). Under rhIL1-β stimulation, however, the availability of the target is strongly increased (Fig. 6B); the wt phage does not bind to hCMEC/D3, even after IL1-β treatment (Fig. 6C). This study demonstrates the specific presence of target molecules on IL1-β-treated endothelial cells for this particular EAE-specific clone; the majority of the tested clones behave in the same manner.

Binding studies of phage clones with endothelial cells in vitro. Confluent hCMEC/D3 cell line was tested (
To further characterize the binding ability of EAE-specific phage clones, protein extracts of confluent endothelial hCMEC/D3 cells, resting or rhIL1-β challenged, were prepared. Clonal phage preparations were tested with both hCEMC/D3 protein extracts in a dot-blot assay. Phage binding was revealed by immunochemistry (Fig. 6D, Table 2). Several of the selected phage clones, characterized by high copy numbers in the EAE and subtraction repertoires but practically absent from the healthy repertoire, reveal strong binding to protein extracts from rhIL1-β-challenged hCMEC/D3 cells compared to the resting condition. Clone N °5, which has high prevalence in the healthy repertoire, binds strongly to the hCMEC/D3 protein extract obtained in the resting culture condition and less to the protein extract of rhIL1-β-challenged hCMEC/D3 cells. A few clones produce background noise-level labeling, similar to that of the wt phage.
Binding scores of phage clones, compared to the wt phage, on protein extracts of hCMEC/D3 cells, stimulated (+) or not (–) with IL1-β, correspond to the optic density (OD) measures of the dark chromogen deposits (see Fig. 6).

Discussion
Without a priori knowledge of target molecules, but with the goal to optimize the peptide biomarker selection of phage-displayed peptides binding to pathological EAE lesions that are disseminated within healthy CNS tissue, we initially performed an in vivo phage-displayed peptide selection of CNS in both EAE-developing and healthy rats. With the emphasis on eliminating the peptides common to both repertoires, we then performed a physical DNA subtraction of these two phage repertoires by DNA hybridization, taking advantage of the circular single-stranded phage DNA (PhiSSH). This process generated a third subtraction repertoire, comprising ȁEAE minus healthy.ȍ The three repertoires were analyzed by NGS, allowing quantitative and qualitative evaluation of the obtained peptides’ DNA-encoding sequences to define a spectrum of peptides that were specifically found in the EAE pathology repertoire.
The aim of PhiSSH is to produce a repertoire enriched in clones specific to a test repertoire (here, the EAE) of phages in order to increase the efficiency of random picking of clones. This is to physically dispose of the material necessary for binding tests on tissues and/or protein extracts. Here, we define the picking efficiency as the probability of recovering various clones specific to the test (EAE) condition.
The NGS data for the three repertoires, such as control (healthy), test (EAE), and PhiSSH (subtraction), allowed us to evaluate the difference of the picking efficiency between the test and PhiSSH repertoires. The theoretical model of the clones of interest is being present in both the test and PhiSSH repertoires, but absent or marginally present in the control repertoire. This can be expressed as 1:0:1, PhiSSH:control:test. In applying this model as the reference matrix, we calculated the Pearson coefficient of the distribution of each peptide in the three repertoires. Thus, we can (a) characterize each clone individually and (b) describe the pertinence of producing and using the PhiSSH repertoire as the sum of the Pearson coefficients for the entire set of clones.
In our experimental work, early attempts to produce a subset of isolated clones specific to the test repertoire were unsuccessful. Indeed, a single clone was being repetitively picked (n = 30). The NGS data explained this observation; in fact, a small subset of clones (n = 18) represents about 47.7% (2,338,654 of 4,903,654) of the test repertoire. During the PhiSSH process, the proportion of these clones has drastically reduced to 1.3% (16,883 of 1,315,957) in the resulting subtraction repertoire.
To optimize the characterization of the PhiSSH repertoire, the theoretical model was adapted to the generated results. By definition, the clones of interest are present in the EAE repertoire (set at 100) and absent from the healthy repertoire (set at 0). By dynamically calculating the sum of the Pearson coefficients while varying the value of the subtraction repertoire in the model (from 70 to 110), we determined the optimal model as 83:0:100 for PhiSSH:control:test (subtraction, healthy, and EAE), using maximization of the sum of the Pearson coefficients as a metric (Supplementary Fig. 1). The resulting distribution of Pearson coefficients was used to determine the cutoff to characterize the individual clones as being of interest or not. Practically, for this particular set of repertoires, a cutoff of 0.631 was used, producing a subset of 72,267 unique peptides present in the PhiSSH repertoire, corresponding to 1,250,769 clones, ~83% of the PhiSSH repertoire (1,250,769/1,510,340 clones of interest/total clones of the repertoire).
These clones correspond to ~97% (4,823,147/4,969,735) of the test repertoire. The apparent picking efficiency seems to be reduced if we do not take into account the overrepresentation of the abovementioned 18 clones. If we do not consider these clones and focus on the ability to randomly pick any other clone, the PhiSSH (subtraction) and test (EAE) repertoires have corresponding efficiencies of ~82% (1,232,437/1,510,340) and ~14% (671,992/4,969,735), respectively. This represents an increase in the picking efficiency of about six times (82/14).
In experimental practice, random picking in the PhiSSH repertoire produced a set of 37 unique peptides (of some 200 picked clones). They were characterized for their ability to recognize EAE lesions on CNS tissues (Fig. 5) and, in a more convenient and cheaper test, for their differential binding on cultured endothelial cells and their protein extracts under resting vs. IL1-β-challenged conditions (Fig. 6). These experimental studies confirmed the ability of several EAE-specific clones to bind preferentially on cultured, IL1-β-challenged, endothelial cells vs. resting ones.
In vivo selection of phage-displayed peptides has been widely used to identify one or a few peptides, preferentially if not specifically, by homing essentially to the luminal surface of blood vessels, with specificities toward organs,4,16 organelles of mammalian cells, 17 or pathological tissues. 18 Well-differentiated target tissues, with important alterations of the vascular compartment identified by peptides, 19 did facilitate the application of the technique to identify vascular targeting peptides. Furthermore, as phage-displayed selected peptides mimic pathological marker proteins that generate autoantibodies, they could serve as marker and follow-up tools for disease evaluation. 20 In these experiments, the overrepresentation of a few specific clones in the test repertoire makes such tasks relatively easy.
In our case, we are seeking to establish the largest possible set of peptides, specifically labeling EAE lesions. However, in neuroinflammatory pathologies such as MS models, disease-affected and healthy tissues are located in close proximity, and the CNS lesions in the animal present different stages of maturation even when first clinical symptoms appear. Therefore, the EAE repertoire (a) is heavily contaminated with peptides targeting sites in the healthy tissues and (b) contains a few overrepresented clones (n = 18) that make the recovery of lower abundance EAE-specific clones practically impossible; even among these 18 clones, one accounts for ~25% of their occurrences, dominating the random picking.
To face the challenge of biomarker discovery in such conditions, we therefore developed a new PhiSSH subtractive approach of healthy and pathological phage-displayed peptide repertoires. The design of our subtractive hybridization procedure allows for the equalization of highly abundant clones for the test repertoire, while those that are abundant and common with the control repertoire are subtracted. There remains one limiting factor for the subtraction efficiency. The single-stranded circular genomic DNA of the phage, materializing the test repertoire, is extremely sensitive to hydrolysis at the applied high temperature to destabilize heterohybrids. To preserve enough material for the recovery of the subtraction repertoire, we limited the reaction time at two hours, thus achieving a relatively low C0t value. While this condition is unfavorable for the subtraction of low-abundance clones that are common with the healthy repertoire, it produces a twofold enrichment of abundant clones across a wide range of frequencies in the EAE repertoire.
The NGS protocol, where a very large number of random DNA fragments are picked and sequenced, reflects large-scale random picking from plaques formed by individual phage clones on petri dishes. The collected NGS data helped to evaluate the efficiency of the PhiSSH protocol and may be helpful to evaluate the complexity of isolated clones that one may achieve for a given number of picked clones and to monitor the random picking process.
In application to neuroinflammation in EAE pathology mimicking major neuropathological aspects of MS, in vivo phage-displayed peptide selection upon injection into blood circulation was evidenced, being particularly adapted to reveal vascular alterations of the BBB. In EAE pathology, opening of the BBB occurs, resulting in focal neuroinflammation lesions characterized by perivascular infiltrates of circulating blood immune cells21,22 involving specific inflammatory molecular expression patterns of endothelial cells. In histopathology, several of the peptides displaying phage clones that were identified as EAE specific revealed vascular binding within the CNS parenchyma of EAE-developing rats, but not with the blood vessels of CNS tissue from healthy control animals. In particular, binding of EAE-specific phage clones to vessels that present characteristic EAE immune cell infiltrates was evidenced. In such neuroinflammation conditions, endothelial cells at the BBB undergo a wide spectrum of molecular and cellular alterations 22 and the set of labeling peptides that we need to observe such changes should be as large as possible. Studies limited to a few proteins, for which reagents are available, often fail to reveal the endothelial cell alterations. With a large set of EAE-specific peptides, we expect to obtain a better description of the time course of these alterations and of the conditions producing such changes by using BBB in vitro models, such as the one used by Weksler et al. 23 , to characterize our first subset of randomly picked peptides from the subtraction repertoire.
The present design of phage display and the PhiSSH for high-throughput screening in peptide biomarker discovery is based on the differential analysis of two repertoires corresponding to two different physiological conditions (healthy vs. pathological). The combined approach could be applied to many diseases presenting disseminated characteristics, such as Alzheimer's or metastasis detection.
Author Contributions
Conceived and designed the experiments: AV, KGP. Analyzed the data: KV-S, AV, KGP. Wrote the first draft of the manuscript: AV, KGP. Contributed to the writing of the manuscript: KV-S, AV, KGP. Agree with manuscript results and conclusions: KV-S, AV, KGP. Jointly developed the structure and arguments for the paper: AV, KGP. Made critical revisions and approved final version: KV-S, AV, KGP. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgments
NGS was performed by the team of the sequencing facility Plateforme de Génomique Fonctionnelle de Bordeaux, the University of Bordeaux. Microscopic examination was done in the Bordeaux Imaging Center of CNRS-INSERM and Bordeaux University. The help of C. Poujol and D. Zaharia is acknowledged.