
Abstract
To determine if candidate cancer biomarkers have utility in a clinical setting, validation using immunohistochemical methods is typically done. Most analyses of such data have not incorporated the multivariate nature of the staining profiles. In this article, we consider modelling such data using recently developed ideas from the machine learning community. In particular, we consider the joint goals of feature selection and classification. We develop estimation procedures for the analysis of immunohistochemical profiles using the least absolute selection and shrinkage operator. These lead to novel and flexible models and algorithms for the analysis of compositional data. The techniques are illustrated using data from a cancer biomarker study.
Introduction
The development of high-throughput assays such as mass spectrometry and gene expression microarrays has led to the generation of large numbers of candidate biomarkers in current medical research. However, while such results and signatures tend to provide great potential for disease prognosis, translating the discovery into a clinically useful biomarker requires more investigation. An important first step typically is to validate the finding using so-called immunohistochemical staining patterns.
In immunohistochemical studies, staining patterns of the biomarker are measured across a variety of samples. One common way this is done is using a tissue microarray 1 In this scheme, the “spots” on the glass slide represent tumor cores from different patients, and an antibody for the protein of interest is applied to the slide. The staining patterns are then correlated with patient characteristics. In most instances, the staining is assessed by a pathologist, who assigns a score on an ordinal scale, with larger values corresponding to higher levels of staining. Note that this structure is quite different from an expression microarray, in which the spots are individual genes and proteins of interest, while what is hybridized to the slide is a single sample.
Typically, an analysis of such data requires the creation of a univariate score measuring staining intensity for each sample. Then the score is associated with clinical outcomes using standard testing and regression methods. For example, if the clinical outcome is binary, one could use a parametric or non-parametric two-sample test for association. Alternatively, one could fit a linear regression of staining intensity on clinical outcome or a logistic regression of the outcome on staining intensity.
As noted by Etzioni et al 2 the staining of tumor samples to the antibody is not completely homogeneous. What is available for certain scoring systems is a multivariate profile of percent of tumor cells staining at each level of the various scoring categories. As an example, we consider tissue microarray data from a candidate prostate cancer biomarker, LIMK1. 3 LIMK1 is a dual specificity novel serine/threonine kinase which modulates actin dynamics through inactivation of the actin depolymerizing protein cofilin.
The function of LIMK1 in reorganization of the cytoskeleton has been studied extensively during developmental defects.4,5 Recently, a role of LIMK1 in progression and invasiveness of breast and prostate cancer has been predicted.6,7 In this paper, we explore the status of LIMK1 staining in the nucleus and cytoplasm as it relates to aggressiveness of prostate cancer.
Summary of staining data for five randomly chosen observations from prostate cancer data.

The structure of this article is as follows. In
Results and Discussion
Data Structures and Logistic Normal Model
We will be assuming that we have data (Di,
Logistic Normal Distribution for Compositional Profiles
Define the (p – 1)-dimensional vector

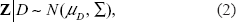
In terms of analyzing immunohistochemical profiles, Etzioni et al
2
adopted a hierarchical model in which priors were placed on μ
D
and σ. They then used a Markov Chain Monte Carlo (MCMC) sampling algorithm to sample from the the posterior distribution of μ
D
. It was used to construct a 95% credible interval for mean shifts for the log-transformed profile. An easier estimation procedure that does not require implementing a Gibbs sampling algorithm is to fit a linear discriminant analysis model to the transformed data
One also notes that (2) can be generalized to allow for proportional covariance matrices across populations. This would then necessitate fitting a quadratic linear discriminant analysis model to
Lasso Estimation
Shifting gears, we discuss the Least Absolute Shrinkage and Selection (LASSO) algorithm proposed by Tibshirani.
10
Suppose we wished to fit the linear regression model: E(Ui|
LIMK1 Biomarker Study
Using the procedures described above as well as in the Methods section, we now consider a real-life application. The immunohistochemical data come from a putative prostate cancer biomarker, LIM kinase 1 (LIMK1). 3 In this study, the expression profile of LIMK1 was determined using a prostate tumor tissue array comprising 50 samples from tumors at different stages of progression. The pool of samples in the array included three uninvolved prostate tissues for comparison. TNM classification of tumors in the TMA indicated that 62% patients had histories of either lymph node or distant metastasis at the time of surgery or biopsy, and 88% of the tumors had Gleason scores of 7 or above. Gleason score (GS) is an aggregate measure of the aggressiveness of the tumor. It is composed of a major and minor Gleason score, each of which is scored on a scale of one to five. We dichotomized Gleason score as less than or greater than or equal to eight.
Analyses of nuclear staining are considered first. Scatterplots of the multivariate nuclear staining profiles by pairwise category comparison are given in Figure 1. To associate staining with the clinical parameters (presence of metastases, Gleason score), we used the product score, multiplying the percentage staining by the staining intensity. Boxplots of the product score for nuclear staining versus presence of metastases and Gleason score are provided in Figures 2 and 3. While Figure 2 indicates that metastatic tumors have higher nuclear staining relative to non-metastatic tumors, there is less difference in nuclear staining across the different Gleason score categories. A t-test reveals the differences corresponding to the boxplot in Figure 2 to be statistically non-significant (P = 0.32 for presence of metastases), while an analysis of variance yields the association between nuclear staining and Gleason score to also be nonsignificant (P = 0.31).
Pairwise plots of nuclear staining by staining category for the LIMK1 study. Boxplot of product score for LIMK1 nuclear staining (vertical axis) by presence of metastases (horizontal axis). 0 indicates absence of metastases, while 1 indicates presence of metastases. Boxplot of product score for LIMK1 nuclear staining (vertical axis) by Gleason score (horizontal axis).


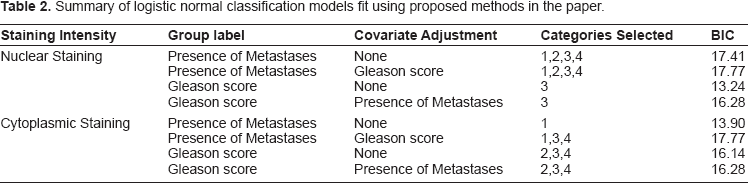
A series of logistic classification models using the proposed methods were run; they are summarized in Table 1. Based on the analyses we find that if we use nuclear staining profile to predict presence of metastases, then all four staining categories are informative. Using the BIC criterion, there was no improvement by including presence of metastases as a covariate. On the other hand, if one wishes to use nuclear staining to predict Gleason score, then only the third staining category is informative. There is no improvement by including presence of metastases as a covariate.
Next, we considered analyses based on cytoplasmic staining intensity. Pairwise scatter-plots of cytoplasmic staining are given in Figure 4. As with nuclear staining, we used the product score for associating the immunohistochemcial profile for cytoplasmic staining with Gleason score and presence of metastases. The boxplots of the LIMK1 cytoplasmic staining product score by presence of metastases and Gleason status are given in Figures 5 and 6. Analyses analogous to those for nuclear staining reveal nonsignificant associations (P = 0.75 and P = 0.40 for presence of metastases and Gleason score, respectively).
Pairwise plots of cytoplasmic staining by staining category for the LIMK1 study. Boxplot of product score for LIMK1 cytoplasmic staining (vertical axis) by presence of metastases (horizontal axis). 0 indicates absence of metastases, while 1 indicates presence of metastases. Boxplot of product score for LIMK1 cytoplasmic staining (vertical axis) by Gleason score (horizontal axis).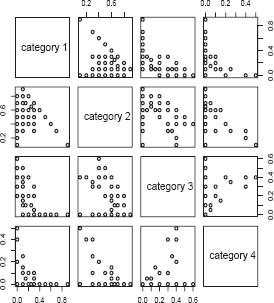


Summary of logistic normal classification models fit using proposed methods in the paper.
Conclusions
In this article, we have explored a compositional data model initially proposed by Etzioni et al 2 that is applicable to the modelling of immunohistochemical biomarker data such as those which might arise from tissue microarrays. We have shown that it has a natural link with linear discriminant analysis, which has been very well-studied in the statistical literature. Consequently, the Etzioni et al model can be fit using standard software packages for LDA, after some data manipulations are performed. The inference we perform is non-Bayesian, in contrast to the Bayesian inference done by Etzioni et al. 2
We also have developed an automated variable selection procedure within the class of models by incorporating LASSO estimation procedures. The real data example shows that this automated variable selection leads to a better fit. We have also outlined a model selection strategy in which the Etzioni et al model is compared to submodels in which categories are suppressed.
While we dealt with the situation in this paper where
Scientifically, while a score-based method such as the product score provides a simple summary statistic for staining data that can then be associated with clinical parameters in tests of hypotheses and regression models, it might oversimplify the data too much. This would be especially undesirable if there is substantial within-sample staining heterogeneity. Thus, methods which explicitly account for the multivariate nature of the staining offer a useful alternative. Compositional data methods are one type of multivariate approach. What our method allows the analyst to do is (1) model the staining profiles in a multivariate manner, (2) incorporate clinical variables as covariates and (3) exclude uninformative staining categories.
Methods
Lasso-Based Optimal Scoring Algorithm
In this section, we describe our proposal, which entails developing a sparse estimator in the logistic normal model for compositional data. This is done by using the optimal scoring algorithm of Hastie et al
11
to convert the logistic normal classification problem into a regression problem. This is done in the following way:
Choose an initial score matrix Fit a linear regression model of M0 on Obtain the eigenvector matrix Φ of Φ of
The fitted values obtained at the end of the algorithm are proportional to the linear discriminant analysis coefficients. To extend the algorithm so that we jointly perform classification and automated variable selection, we simply replace step 3 of the algorithm by LASSO estimation of the type described in Results and Discussion. Based on the algorithm, regression coefficients for each variable in
We will use the algorithm of Osborne et al
13
for LASSO estimation. Let σ be the index set, a subset of {1, …, p}. The ith component of β is non-zero if and only if I ∊ σ. The algorithm of Osborne et al
13
operates by sequentially updating the index set. Let P denote the permutation matrix that arranges the non-zero components of η as the first s components, where s is the cardinality of σ. We have that

Let Θσ be the sign vector of βσ. At each step of the algorithm, β must satisfy the L1 constraint; this can be expressed as 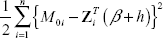
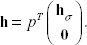
If the constraint is active, then the optimal solution for Find the smallest α ∊ (0, 1) such that 0 = β
k
+ αhk for a k ∊ σ and set One of two steps may be taken here. Either
Set Θ
k
= –Θ
k
and recompute Update σ by deleting k, resetting β and Θσ accordingly, and recompute Iterate between steps 1 and 2 until a sign feasible
Once the sign feasibility is obtained, the optimality of the candidate solution is tested. This is done by calculating
Find the index j such that the jth component of v2 has the largest magnitude. Update σ by adding j to it and update βσ by adding a zero as its last element and βσ by appending the jth component of sign(v2). Set (β* = β and iterate between steps 1 and 2.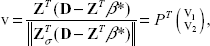
This algorithm has been implemented as an R function (www.r-project.org) by the first author and can be obtained upon request.
Covariate Adjustment and Model Selection
An important question not addressed by Etzioni et al
2
was adjusting for other co-variates in addition to D. Since we have expressed the classification problem as a regression one via the optimal scoring algorithm, we can immediately modify it to account for Choose an initial score matrix Fit a linear regression model of Compute the residuals from step 2 and regress on Obtain the eigenvector matrix Φ of
Notice that this algorithm forces
Based on the models, it would be useful to have a criterion for performing model selection. We can do this easily again using the equivalence of the classification and regression problem. We simply use the formula RSS + p/2 log n, where RSS denotes the residual sum of squares from the linear regression output in the algorithm, and p denotes the number of variables that are in the regression model. In particular, variables with estimated zero coefficients are not counted. Lower values of the criterion indicate better model fit. We will refer to this criterion as the Bayesian Information Criterion (BIC), a version of which was proposed by -Schwarz. 14 Note that if the smallest BIC value corresponds to no variables being excluded, then this indicates that the best model fit is that of Etzioni et al. 2
Disclosure
The authors report no conflicts of interest.
Footnotes
Acknowledgments
The first author is supported by NIH R01-CA129402. The first author would like to acknowledge discussions with James MacDonald. The second author is supported by the Prostate Cancer Research Program of the Department of Defense (PCRP PC041048-RC).