
Abstract
Significant advances in our knowledge of cancer genomes are rapidly changing the way we think about tumor biology and the heterogeneity of cancer. Recent successes in genomically-guided treatment approaches accompanied by more sophisticated sequencing techniques have paved the way for deeper investigation into the landscape of genomic rearrangements in cancer. While considerable research on solid tumors has focused on point mutations that directly alter the coding sequence of key genes, far less is known about the role of somatic rearrangements. With many recurring alterations observed across tumor types, there is an obvious need for functional characterization of these genomic biomarkers in order to understand their relevance to tumor biology, therapy, and prognosis. As personalized therapy approaches are turning toward genomic alterations for answers, these biomarkers will become increasingly relevant to the practice of precision medicine. This review discusses the emerging role of genomic rearrangements in breast cancer, with a particular focus on fusion genes. In addition, it raises several key questions on the therapeutic value of such rearrangements and provides a framework to evaluate their significance as predictive and prognostic biomarkers.
Introduction
The role of chromosomal rearrangements in tumorigenesis and cancer progression has received substantial attention in recent years.1–3 Chromosomal rearrangements were initially described in hematologic malignancies such as chronic myelogenous leukemia (CML)4,5 and Burkitt's lymphoma,
6
where they have been used both for diagnosis and to direct targeted therapies. Subsequently, recurrent translocations were also found in rare classes of soft tissue tumors such as Ewing's sarcoma7,8 and synovial sarcoma.
9
Recurrent translocations were not initially identified in many of the common solid, epithelial tumors, in part, because of the limitations of standard cytogenetic analyses and the underlying biological diversity. However, with the emergence of new technologies that allow more comprehensive genomic analysis of solid cancers, genomic rearrangements have been identified in many solid tumors, including breast cancer. This has enabled the identification of subsets of common solid tumors that harbor novel fusions or rearrangements that were not previously appreciated, eg,
Tumor genomic profiling encompasses a variety of sequencing techniques that use next-generation sequencing methods, eg, DNA and RNA sequencing (RNA-seq). Genomically-guided therapy or targeted therapy refers to the selection of a treatment strategy based on the results of tumor genomic profiling, where a clinical response is more likely to occur in the presence of the relevant genomic target. Association between the presence of a genomic alteration and drug response defines a genomic alteration as a predictive biomarker. Such biomarkers have been critical to personalizing the approach to cancer treatment and improving patient outcomes. In contrast, prognostic biomarkers define disease trajectory in the untreated individual. Although some biomarkers can be both predictive and prognostic, biomarkers that are only prognostic can be useful in defining subsets of patients at risk for poor outcomes. Such knowledge allows the treating physicians to determine whether more aggressive or alternative approaches should be undertaken for those patients. Many genomic alterations, including point mutations, deletions/insertions, amplifications, and rearrangements, serve as predictive biomarkers, prognostic biomarkers, or both.
Genomic rearrangements refer to structural changes in the genome that are caused by breakage of DNA followed by erroneous rejoining and replication. These include events that alter copy number, such as deletion, tandem duplication, and amplification, as well as others that maintain copy number, such as reciprocal translocations and inversions (Fig. 1A–C). Rearrangements encompass gross alterations of the whole chromosome or part of a chromosome and do not include the more commonly studied single base mutations or small deletions and insertions of a few base pairs in length. A special class of rearrangements known as interchomosomal or intrachromosomal rearrangements is the result of interactions between distant regions of the genome or even within the same chromosome, respectively. This type of rearrangement can lead to fusion of two disrupted genes, resulting in an altered transcript and a fusion protein (Fig. 1B). These fusions can potentially activate, reduce, or eliminate the original function of the gene product(s) or generate a chimeric protein. Neomorphic functions may also result and have been described, eg, gain of function
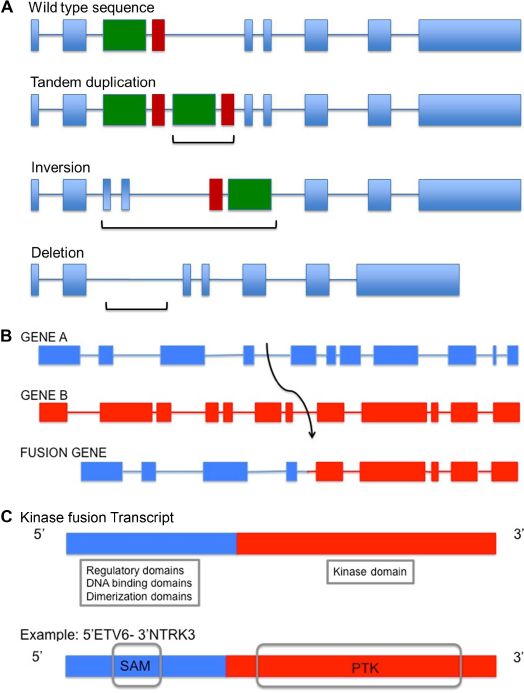
Illustration of genomic rearrangements and gene fusions. (
Detection of recurrent genomic alterations provides new prognostic biomarkers, enables selection of patient groups that may most benefit from specific targeted agents, predicts their response to targeted therapy, and affords the opportunity to elucidate both intrinsic, tissue-specific and acquired resistance mechanisms. With the advent of personalized medicine in cancer, the need for comprehensive genomic profiling of difficult-to-treat tumors is becoming more apparent. While a wealth of information is being generated in the process, characterizing biomarkers for patient classification, prognosis, predicting drug response, and resistance to treatment is crucial.
Defining prognostic and predictive biomarkers in breast cancer is more complicated than in other tumor types. This is primarily because breast cancer represents a heterogeneous set of diseases with distinct molecular features, natural course of disease, and response to treatment. Recognition of this heterogeneity in more recent studies has allowed more precise understanding of molecular characteristics that influence drug response and patient outcomes. Breast cancers are clinically subtyped based on three biomarkers: expression of estrogen receptor (ER) and progesterone receptor (PR) as assayed by immunohistochemistry (IHC) and expression of human epidermal growth factor receptor 2 (HER2) or amplification of erb b2 receptor tyrosine kinase 2 (
Genomic profiling using high-throughput, next-generation sequencing technologies (ie, whole-exome, whole-genome, and RNA-seq) has identified recurrent point mutations in breast cancer subtypes:
In this review, the current knowledge of chromosomal instability (CIN) in breast cancer and implications of prognosis for the different molecular subtypes is explored. The patterns and frequency of genomic rearrangements in breast cancer are also discussed. Since more recent knowledge on genomic rearrangements relies heavily on the technique used to study them, the most relevant roles of different technologies and the information acquired are described. Given the therapeutic potential of fusions in cancer, the benefits of identifying and characterizing such rearrangements in breast cancer is outlined. Finally, the current knowledge of breast cancer-related fusions as predictive biomarkers, the future of this evolving field, and the clinical potential for improving therapeutic options for patients with breast cancer is discussed.
Chromosomal Instability and Breast Cancer Prognosis
Genomic rearrangements are closely associated with CIN, which is defined as a dynamic state in which gains or losses of whole or parts of chromosomes occur. Such instability can alter the number of chromosomes, a phenomenon known as aneuploidy. Some cancers such as breast and colorectal cancers harbor more CIN as compared to others.23–25 Though the mechanism of CIN is poorly understood, the implications of its extent have been investigated in relation to clinical outcomes in breast cancer subtypes.
Several groups have analyzed the overall patterns of CIN of breast cancer subtypes and the relevance to clinical outcomes (Table 1). In these studies, CIN was measured based on DNA copy number changes and losses or gains of chromosomal regions or chromosomal number changes in tumor nuclei.26–28 To evaluate the prognosis in some cases, data were retrospectively compared to the clinicopathological details of treatment-naïve patients. CIN is generally considered to be associated with poor prognosis in solid tumors.29,30 While analysis between ER+ and ER– subtypes confirmed that this is true for ER+ cancers, extreme CIN did not show a clear association with survival outcome or prognosis in ER– cancers.26,31 Another study evaluating CIN on the basis of copy number changes found a correlation between increased CIN and poor survival outcomes in ER+ and HER2+ subtypes. 27 Similar to the previous reports, CIN score was higher in ER- and TNBC samples. However, there was no correlation with survival outcome in these tumors. Major differences between the studies, including small sample size within CIN study cohorts, different methods for evaluating CIN, presence of confounders, absence of detailed patient and treatment profiles, and other parameters, make interpretation and comparison of such data difficult. These studies highlight the complexity of breast cancer genomes and point to the fact that while instability might be used to assess risk in ER+ cases, additional distinct biological markers for predicting clinical outcome in ER- cases are needed.
Association between chromosomal instability and outcomes in breast cancer subtypes.
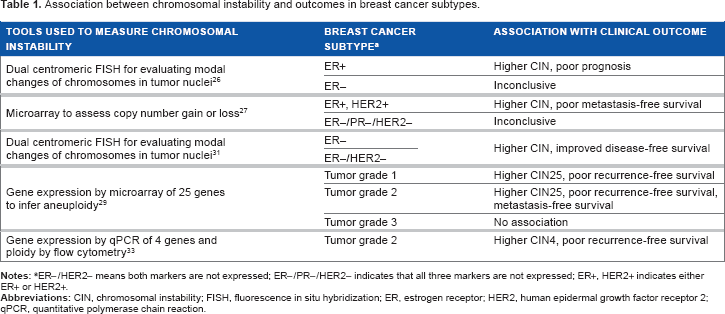
ER-/HER2- means both markers are not expressed; ER-/PR-/HER2- indicates that all three markers are not expressed; ER+, HER2+ indicates either ER+ or HER2+.
Other groups have developed methods to identify gene expression signatures that reflect CIN and analyze associations with relapse, 32 prognosis, 33 and survival outcomes 29 (Table 1). Tumor grade, which reflects the differentiation and proliferation potential of tumor cells, is a routinely used histological measure. Based on the expression of the top 25 genes in a 25-gene expression assay known as CIN25, grade 1 and grade 2 breast tumors from three data sets were stratified to high or low scores. 29 The genes within the panel were selected based on strong associations between altered gene expression and tumor aneuploidy. A higher CIN25 score was associated with a worse clinical outcome for patients with either grade 1 or grade 2 tumors. Similarly, another study using only four genes (CIN4) found a significant association with the proliferation marker Ki67 in grade 2 tumors. CIN4 was used to further stratify patients with grade 2 tumors into good and poor prognosis groups. 33 Higher CIN4 was associated with worse recurrence-free survival. Data from these studies, summarized in Table 1, highlight the differences in CIN and the outcomes observed between breast cancer subtypes.
Patterns and Frequency of Genomic Rearrangements in Breast Cancer
An increase in genomic instability has been linked to a concomitant increase in the frequency of gene rearrangements or fusions.2,34 Next-generation sequencing strategies have led to genomic and transcriptomic analysis of large cohorts across cancer types as well as detailed analysis of the patterns of genomic rearrangements in breast cancer.34–36 Some breast cancers showed genome-wide rearrangements, whereas others were reported as clusters in regions of amplification. Array-based comparative genomic hybridization (array CGH) studies of copy number alterations have previously defined structural changes in the genome in terms of gains or losses of specific chromosomal regions such as 1q/16 for low-grade ER+ tumors, commonly amplified sites such as 8p11–12 (
Using next-generation technologies, quantification of the number of rearrangements occurring within chromosomes (intrachromosomal rearrangements such as duplications, inversions, amplifications, and deletions) and also those occurring between different chromosomes (interchromosomal rearrangements) has been depicted by Kwei et al 39 using circos plots for breast cancer. Circos plots are circular illustrations for visualizing the structural relationships between regions of chromosomes. These comparisons clearly show distinct patterns related to breast cancer subtypes. 39 Low-grade ER+ breast tumors generally display few rearrangements and amplifications, whereas high-grade ER+ breast cancers and TNBCs display a large amount of both large- and small-scale rearrangements, especially increased frequency of intrachromosomal rearrangements such as tandem duplications in TNBCs. This implies that subtype-specific differences in genetic instability may mechanistically contribute to different gene expression patterns observed in breast cancer subtypes.36,39,43
By analyzing 24 breast cancer genomes by paired-end sequencing, Stephens et al
36
showed that intrachromosomal alterations are much more prevalent than anticipated across a broad spectrum of molecular subtypes based on ER/PR/
Although it has been shown that tandem duplications are the most commonly observed rearrangement in breast cancer genomes, the frequency of specific tandem duplications is currently unknown as these alterations are not easily detected with standard techniques such as array CGH or FISH. Next-generation sequencing techniques will achieve improved clarification of the genes and gene regions that are frequently involved in such duplications.
Role of Technological Advances in Identifying Genomic Rearrangements
The advantages of newer methods to study breast cancer genome have resulted in a greater understanding of the patterns of genomic instability and the underlying gene rearrangements observed within breast cancer (Table 2). Some of these methods include FISH, break-apart FISH, array CGH, polymerase chain reaction (PCR)-based techniques, whole-exome sequencing, whole-genome sequencing (WGS), RNA-seq, single primer enrichment technology (SPET), and anchored multiplex PCR (AMP).
Comparison of methods used to identify rearrangements.
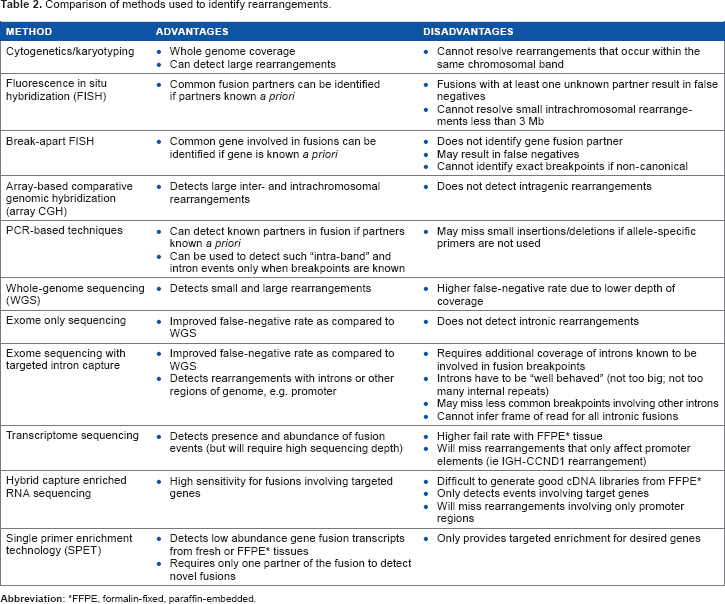
FFPE, formalin-fixed, paraffin-embedded.
FISH is a powerful tool for detecting specific genomic alterations and has gained popularity due to its clinical application in identifying
The development of more efficient high-throughput sequencing methods and data analysis pipelines has made the identification of pathogenic rearrangement events across all malignancies more affordable and more accessible. WGS and RNA-seq provide an unbiased view of the genome and expressed transcripts, respectively (Table 2). A subset of these next-generation sequencing methods is designed to capture rearrangement events in particular, eg, intron capture and RNA-Seq. Using WGS and RNA-seq, Stephens et al 36 showed that multiple rearrangements are present in many breast cancers, with .50% of them occurring within coding regions. 36 These rearrangements can lead to deregulation of gene expression and/or the formation of fusion transcripts, resulting in novel fusion proteins. 49
To detect rearrangements, read pairs with unexpected separation distances or orientations that discordantly map to two distinct genes are identified. In order to determine the exact position of the breakpoint, reads that partially map to both genes are then reviewed. Carrara et al 50 have comprehensively reviewed the RNA-seq fusion detection tools, and Lin et al 51 have detailed the WGS statistical algorithms that detect such variations (structural variation callers). High-throughput sequencing technologies suffer from greater error and shorter reads than traditional sequencing methods, and the bioinformatic pipelines often result in a long list of fusion candidates that require extensive experimental validation. 52 Advanced computational analyses are, therefore, required to decipher true rearrangements from false positives that are due to reverse transcriptase template switching, incorrect mapping, read-through transcripts from the splicing of two adjacent genes, and other systematic errors. Further analysis can also help in determining the pathogenic significance of these candidates.53,54 These methodologies have led to the discovery of important fusion events in hematologic and solid tumors.55–57
Whole-exome sequencing can identify rearrangement events whose fusion junctions also occur in the coding region.56,58 More recently, deep sequencing platforms have been developed that capture introns and can detect fusions with intronic breakpoints, previously undetected by exome sequencing only.
59
Further, rearrangements occurring in other noncoding regions of the genome can place a gene (or part of it) under the regulation of a different gene promoter, eg,
Despite the increased sensitivity in detecting rare events, massively parallel sequencing is prone to error at many levels such as library preparation, analysis, and referencing due to the vastness of the genome and similarity between genes (Table 2).61,62 Due to the reduced quality of formalin-fixed, paraffin-embedded samples, tumor tissue contamination, and low frequency representation of chimeric transcripts, the reduced number and depth of reads can lead to false-negative results in RNA-seq. Newer methodologies are being developed to combine RNA-seq and WGS data to improve sensitivity and specificity. 63 For a more specific and reliable detection of fusions, SPET and AMP are currently being explored.64,65
Clinical Significance of Fusions in Cancer
Gene fusions observed repeatedly in certain tumor types are referred to as recurrent gene fusions. Though previously underappreciated in comparison to hematologic malignancies, more recently discovered recurrent gene fusions have been described in solid cancers, including
A recent study using RNA-seq, DNA copy number analysis, and gene mutation analysis of ~4000 primary tumors showed that tumors harboring transcript fusions had significantly fewer driver mutations, suggesting a tumorigenic role for gene fusions. 34 Tumors with recurrent, in-frame fusion transcripts were found to have reduced number of gene mutations in comparison to those without recurrent in-frame fusions. This was a common finding in many cancers types, including breast cancer, suggesting that fusions can drive cancer growth and progression in epithelial tumors.
It is important to note that rearrangements not only create novel driver oncogenes but can also disable critical tumor suppressors. In a recently published study, it was shown that ~16% of the osteosarcomas, which lacked commonly known hotspot
Recurrent gene fusions represent a unique class of rearrangements that can serve as predictive or prognostic biomarkers or both. The most commonly observed gene classes involved in fusions are kinases and transcription factors, together representing >50% of all genes found in fusions.
73
Recurrent fusions that result in the activation of tyrosine kinases such as
Examples for kinase fusions with multiple 5′ partners, relevant inhibitors, and reported tumor tissue types.
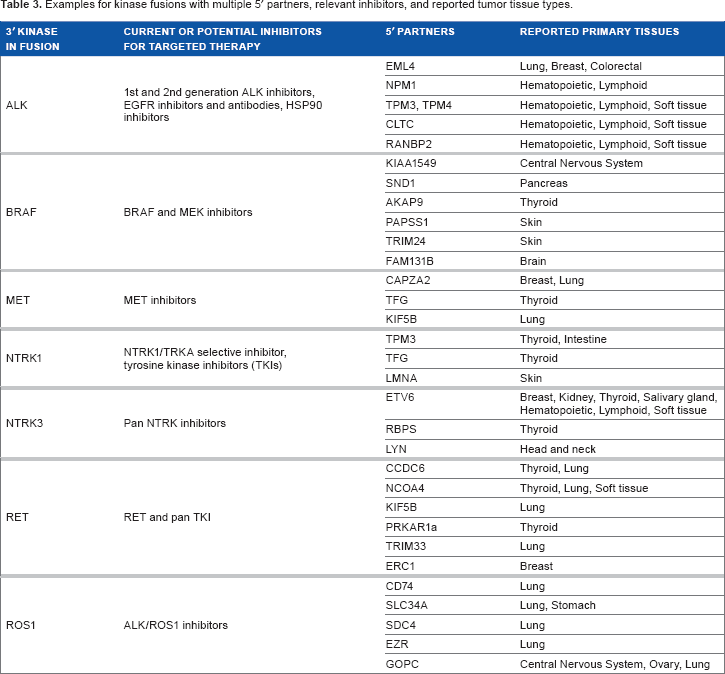
While some fusions serve only as predictive biomarkers for RTKi therapy, others may play a role both as predictive and prognostic biomarkers. Exemplifying this concept is the recurrent fusion between
In contrast to kinase fusions that most often result in kinase activation, transcription factor fusions can lead to gene activation or to altered gene expression affecting integral mediators of cell function. This can have a dominant negative effect on the cell. In follicular thyroid cancers with
Approximately 50% of prostate cancer cases are known to harbor fusions in the Ets transcription factor family with TMPRSS2, a serine protease.66,73,81 Almost 80% of these fusions are
Due to their role, recurrence, and functional implications, fusions thus serve as powerful, predictive biomarkers for targeted therapy, have prognostic implications, and display significant translational relevance.
Current Knowledge of Fusions in Breast Cancer
Large-scale studies involving transcriptome and genomic sequencing have revealed the presence of several gene fusions in the more common types of breast cancer.3,34,36 These include fusions that have been noted in other cancers (Tables 3 and 4). Though several in-frame fusions were observed, a recurrent gene fusion is yet to be identified.3,36 With a few exceptions, the majority of fusions reported in breast cancer are uncommon and present only in a limited number of samples. Whether these are truly single events or are not observed in more samples due to limited sample size, difference in specific breast cancer subtypes included in study cohorts, and/or the appropriate technologies for detection is yet to be determined.
Therapeutic implications for fusions reported in breast cancer.
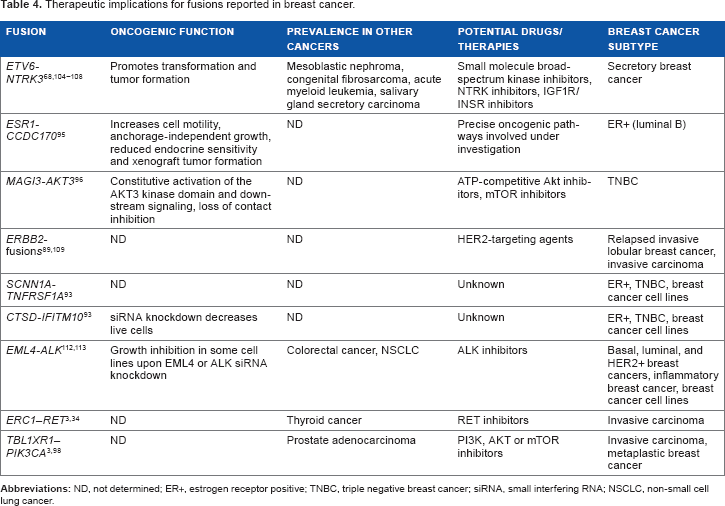
Thompson et al
89
recently reported fusion transcript analysis by five different groups from 813 breast tumors from The Cancer Genome Atlas (TCGA). Although the presence of specific recurrent fusion transcripts was low, most tumors were reported to have at least one or more fusions.
Several groups have identified recurrent fusions with potential clinical implications. Robinson et al
92
identified the presence of several recurrent gene rearrangements involving
Enrichment of some fusions may also reflect specific molecular subsets of breast cancer. For instance, an integrative pipeline to probe TCGA revealed
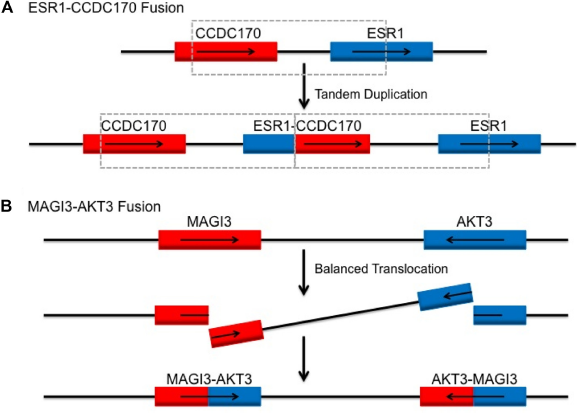
Genetic mechanism for reported recurrent fusions
Other breast cancer subtypes with distinct biological behavior have been genomically evaluated using next-generation sequencing strategies. One study that sequenced TNBC samples in Mexican and Vietnamese populations reported ~7% recurrence of
Metaplastic breast cancer is a particularly aggressive form of TNBC affecting mesenchymal breast cells. Because it does not respond to standard therapies, the prognosis is worse for patients with this breast cancer subtype as compared to other TNBCs. Characterization of a small cohort (
An analysis of relapsed invasive lobular breast cancers reported the presence of a novel
As this is a rapidly evolving field and new information is cataloged daily, some useful websites and data portals for gene fusions in breast cancer include those through the National Cancer Institute, 110 Wellcome Trust Sanger Institute, 111 and TCGA fusion gene data portal. 34
Therapeutic Implications of Fusions in Breast Cancer
Adaptation of trial design has become more important, given the growing knowledge of genotype-drug response associations. Tumor-specific trials still have relevance; however, many recurrent genomic alterations, including rearrangements, identified in one cancer subtype are also seen in other cancers. This underscores the need for expanded eligibility criteria for enrolling patients in clinical trials.
As more pan-cancer studies are revealing recurrent fusions across tumor types, the concept of basket clinical trials is now being evaluated where patients are matched based on their genomic alteration rather than solely on the basis of tumor type. Basket trials that expand eligibility of cancers with novel genomic alterations would allow investigation of efficacy of targeted agents for previously unreported alterations. Singh et al
56
reported the discovery of a highly oncogenic fusion protein,
A caveat to basket trials is that rearrangements may not exhibit the predicted response to drugs. Such an example is the
Although the prevalence of fusions in breast cancer has been reported in many studies,3,34,36 much less is known about the role of fusions in breast cancer. Functional evaluation of such genes is critical to understand the scope of therapeutic relevance. Validation of top candidates found in cancer cell lines and tumor tissue cohorts suggest potential oncogenic mechanisms and therapeutic opportunities. Table 4 summarizes some of the fusions reported to be recurrent in breast cancer cell lines and tumor subtypes, their oncogenic characterization, prevalence in other cancers, and potential therapeutic opportunities for each. In the latest precision medicine approach for difficult-to-treat cancers, regardless of the recurrence of fusion genes, even the presence of one or more actionable kinase fusions is applicable for targeted therapy.
Small molecule kinase inhibitors are the most commonly used targeted approach (Table 3). However, kinase fusion partners may give rise to their own functional consequences and have also been exploited for the development of targeted treatment strategies. Use of ABL kinase inhibitors for
Similar to kinase fusions, transcription factor fusions have also been explored for targeted therapy. For the acute promyelocytic leukemia-associated
Future Perspectives
Detection of relevant and actionable genomic alterations is at the forefront of personalized therapeutic practice in cancer. Apart from therapy selection and prognosis, there have been clinical reports indicating the usefulness of rearrangements in providing diagnostic clarity,125,126 investigating mechanisms of acquired drug resistance, 127 and exploring novel combinatorial therapy. High-throughput genomic sequencing studies, such as exome sequencing of human cancer by TCGA and use of limited hotspot panels, have focused on identifying point mutations and rearrangements specifically involving exons. However, intronic rearrangements are also common in many solid tumors, including breast cancer, and may represent a large class of actionable genomic alterations that are missed by standard short-read sequencing approaches. Inexpensive, rapid turnaround, and clinically implementable sequencing approaches that readily identify potentially actionable genomic rearrangements are clearly needed in conjunction with continued characterization of novel fusion genes.
While our understanding of functional rearrangements in breast cancer is emerging, other overarching challenges remain. These challenges include how to ensure the quality and depth of sequencing reads, standardized reporting and validation across studies, tumor sample purity, clonal heterogeneity, and multifocality. Less well-understood transposable elements such as LINE1 and Alu, which have not been addressed in this review, are also being investigated to define their role in genomic instability and cancer.128,129 Newer techniques of genome editing such as CRISPR, which allow precise manipulation of the genome at a desired location, are under study to model genomic alterations more efficiently and also to develop gene therapy.130–132 Nevertheless, as our understanding grows with affordable, but sophisticated, sequencing strategies and metagenomic approaches, rearrangement-based biomarkers will be pivotal for the practice of precision medicine in breast cancer.
Author Contributions
Wrote the first draft of the manuscript: BSP and KMH. Contributed to the writing of the manuscript: BSP, SCD, HK, SG, and KMH. Jointly developed the structure and arguments for the paper: BSP and KMH. Made critical revisions and approved final version: BSP, SCD, HK, LRR, SG, and KMH. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgment
We would like to acknowledge Ms. Jacqueline Harris for her support and assistance.