
Abstract
Sequence alignment is an important tool for describing the relationships between DNA sequences. Many sequence alignment algorithms exist, differing in efficiency, in their models of the sequences, and in the relationship between sequences. The focus of this study is to obtain an optimal alignment between two sequences of biological data, particularly DNA sequences. The algorithm is discussed with particular emphasis on time, speedup, and efficiency optimizations. Parallel programming presents a number of critical challenges to application developers. Today's supercomputer often consists of clusters of SMP nodes. Programming paradigms such as OpenMP and MPI are used to write parallel codes for such architectures. However, the OpenMP programs cannot be scaled for more than a single SMP node. However, programs written in MPI can have more than single SMP nodes. But such a programming paradigm has an overhead of internode communication. In this work, we explore the tradeoffs between using OpenMP and MPI. We demonstrate that the communication overhead incurs significantly even in OpenMP loop execution and increases with the number of cores participating. We also demonstrate a communication model to approximate the overhead from communication in OpenMP loops. Our results are astonishing and interesting to a large variety of input data files. We have developed our own load balancing and cache optimization technique for message passing model. Our experimental results show that our own developed techniques give optimum performance of our parallel algorithm for various sizes of input parameter, such as sequence size and tile size, on a wide variety of multicore architectures.
Introduction
Over the past decade, we have observed an exceptional development in molecular biology.1,2 An extremely high number of organisms have been sequenced in genome projects and included in genomic databases for further analysis. These databases present an exponential growth rate, and they are intensively accessed daily. Biologists are thus faced with the problem of dealing with huge databases with very large sizes of sequences, in the search of meaningful similarities among biological sequences. 3 In order to do that, high computing power and large storage space during computation are necessary. Moreover, sophisticated algorithms that realistically model the relationships among the organisms must be used. Sequence alignment is important in many different areas as a method to infer how two or more sequences are related. Areas such as computer vision, speech recognition, evolutionary biology, and DNA sequencing have seen the use of sequence alignment algorithms. However, this study focuses on the application of sequence alignment algorithm, in particular for nucleotide sequences.
Parallel computation promises shorter execution times and the ability to process greater quantities of data compared to sequential computation. However, in practice, it is hard to realize a parallel implementation that comes close in achieving its theoretical potential. This is because efficient cooperation between processors is difficult to implement. Parallelism introduces a new set of concerns for the programmer: the scheduling of computations, placement of data, synchronization, and communication between processors. This adds greatly to the complexity of the programming task. An implementation must manage all these concerns in addition to computing a result.
Parallel programming languages and methodologies typically attempt to assist the programmer in one of two ways. The first approach is to provide layers of abstraction that hide the low-level details of the parallel machine from the user. This simplifies the programming task but reduces control over the finer details of the parallel implementation. Other languages provide as little abstraction as possible and require the parallelization concerns to be managed explicitly. A skillful programmer can produce efficient implementations in such languages. However, they are hard to use effectively; furthermore, the code produced is often unclear, brittle, and machine specific. In our implementation, we have used different approaches for different memory architectures, such as shared memory and distributed memory.
Nowadays, multicore processors are more popular for commercial and home usage. This is due to the decreasing cost of their production and the physical limitations in the production of classical processors. It becomes increasingly difficult to enhance processor speed; therefore, multiplying processing units seems to be the best way to achieve larger performance. Developing programs for these processors requires some specific knowledge about the processor architecture and parallel programming. On the other hand, different operating systems provide mechanisms that allow efficient use of more than one processing unit, but still the knowledge related to the processor architecture is needed. The next problem that should be solved in this case is the problem of efficient program execution. When the number of program blocks that can be executed in parallel is larger than the number of available processing units, it leads to even a decrease in the performance due to frequent context switches of the working processes. Therefore, the most suitable situation is when the number of program blocks and the number of available processing units are the same. However, this hampers the portability of the program, which means that during its execution on the processor with different number of processing units, the efficiency can be worse. On the other hand, there are available tools that help in program development, for example, SWARM, 4 or execution environment such as RapidMind. 5 It takes serial program as the input and then executes it in parallel using the virtual machine. Both of the above solutions still require some knowledge about the processor architecture and parallel programming. As an alternative solution, the automatic program parallelization can be used. In our implementation during programming multicore processors, the shared memory paradigm uses OpenMP API and the distributed memory paradigm uses OpenMPI libraries. The main focus of research in this work is the parallel computational aspect of the biological algorithms. A new method is developed to reduce the space complexity of a fast standard alignment algorithm for comparing two sequences using gap cost. The proposed techniques were implemented on a cluster of homogeneous and heterogeneous workstations, and efficient parallelization strategies were implemented and compared to accelerate our algorithms used for bioinformatics analysis. The remaining of this paper is structured as follows. The “Methods and materials” section gives the detailed literature survey related to this work. It exemplifies the study of basic bio-based algorithm and various methods that had already been used to solve the particular problem. The “Proposed methods” section provides the description of our proposed methods and work on various architectures carried out for different datasets. The main results in terms of performance measurement of our algorithm are presented in the “Experimental results” section. The “Conclusion” section gives concluding remarks.
Methods and Materials
Dynamic programming in computational biology
Dynamic programming is a computational technique based on the principle of optimality, 6 which states that the optimal solution to a subproblem is part of the optimal solution to a larger problem. Dynamic programming has been used to efficiently solve many combinatorial optimization problems in polynomial space and time. Dynamic programming is widely used to process biosequence information. The recent arrival of high-throughput next-generation sequencing machines 7 allows researchers to generate millions of short sequence reads in parallel at low cost. Our domain of interest is parallelizing computationally expensive portion of biological algorithms using different parallel models and studying the comparative performance on multicore architecture. The dynamic programming technique has been used in computational biology since the early 1970s for pairwise sequence comparison, multiple alignment, and secondary structure prediction. 8 A search for the phrase “dynamic programming” in the Journal of Bioinformatics, BMC Bioinformatics, and IEEE/ACM Transactions on Computational Biology and Bioinformatics returned more than 1,400 articles. The National Center for Biotechnology Information (NCBI) BLAST 9 compares a virus sequence against the NR nucleotide database containing 14,332,765 sequences. The search completed in tens of seconds and returned less than 1,934 matching sequences (<0.14% of the database) sorted by the statistical significance of each match. One way of accelerating search applications is to use heuristics to approximate the dynamic programming computation. Examples include NCBI BLAST 9 and HMMERHEAD 10 to accelerate pairwise sequence alignment and motif finding, respectively. These applications use a variety of computationally inexpensive kernels to discard a majority of inputs before applying expensive dynamic programming.
A bottleneck in computational biology analysis
Before 2005, advances in Sanger sequencing technology, which was used to sequence entire genomes and was primarily responsible for the growth of Swiss-Prot, doubled the number of bases sequenced per dollar every 19 months. In other words, sequencing costs halved in price every 19 months. More strikingly, starting in 2005, there was a marked expansion in DNA sequencing with the arrival of next-generation sequencers (NGS). 9 NGS allow high-throughput sequencing at a fraction of the cost of traditional Sanger sequencing and enabled applications such as re-sequencing of genomes and meta-genomics, which studies the genomes of mixtures of microorganisms in environments such as deep mines, ice cores, sea water, or the human gut. Since the emergence of NGS, the number of bases that can be sequenced per dollar has been doubling every six months as sequencing costs have plummeted. In Ref. 11, the author states that they expect this growth to continue in the near future as companies push toward the goal of a $1,000 sequencing of a single human genome. Our analysis in this section has shown that the speed of an individual dynamic programming-based comparison is advancing at a sluggish pace, primarily because processor frequency has stagnated. It has become necessary to parallelize search applications on multicores to track the Moore's law curve. However, low-cost sequencing is increasing the amount of biosequence data available and is stressing computational pipelines. The advent of NGS in 2005 created an inflection point where sequencing capacity increased by an order of magnitude without a corresponding improvement in computational power. Subsequently, DNA-sequencing capabilities are increasing at a far higher pace than compute performance. We assert that as costs for sequencing continue to fall, compute performance, not sequencing, will become the bottleneck in advancing genome science. We limit our analysis to twin CPU systems, where the twin chip set is placed in one socket of a motherboard. A chip may in turn have multiple cores (in our case, six cores), and each core may have one or more hardware threads.
A brief introduction to DNA sequence alignment
Sequence alignment involves finding similarities between two sequences. To compare two sequences, we need to find the best alignment between them, that is, to place one sequence above the other making clear the correspondence between similar characters. 12 In an alignment, spaces can be inserted in arbitrary locations along the sequences so that they end up with the same size. Given an alignment between two biological sequences s and t, a score can be assigned to it as follows. For each column, we assign, for instance, +1 if the two characters are identical, 0 if the characters are different, and -1 if one of them is a space. The score is the sum of the values computed for each column. The maximal score is the similarity between both sequences. Figure 1 shows the alignment of sequences s and t, with the score for each column. An exact algorithm (NW) 13 based on dynamic programming that obtains the best global alignment between two sequences was proposed by Needleman and Wunsch. 13 To compute exact local sequence alignments, Smith and Waterman 14 proposed an algorithm (SW), also based on dynamic programming, with quadratic time and space complexity. Hirschberg 15 proposed an exact algorithm that calculates a local alignment between two sequences s and t in quadratic time but in linear space. This approach splits sequence s in the middle, generating subsequences s1 and s2, and calculates the corresponding place to cut sequence t, generating subsequences t1 and t2, in such a way that the alignment problem can be solved in a divide and conquer recursive manner. Usually, one given biological sequence is compared against thousands or even millions of sequences that comprise genetic data banks. One of the most important gene repositories is the one that is part of a collaboration that involves GenBank at the NCBI, the EMBL at the European Molecular Biology Laboratory, and DDBJ at the DNA Data Bank of Japan.
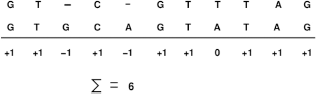
Sequence alignment between s = GACGATTAG and t = GATCGAATAG.
These organizations exchange data daily, and a new release is generated every two months. By now, there are millions of entries composed of billions of nucleotides. In this scenario, the use of exact methods such as NW and SW is prohibitive. For this reason, faster heuristic methods are proposed, which do not guarantee that the best alignment will be produced. Usually, these heuristic methods are evaluated using the concepts of sensitivity and sensibility. Sensitivity is the ability to recognize as many significant alignments as possible, including distantly related sequences. Sensibility is the ability to narrow the search in order to discard false positives. 16 Typically, there is a tradeoff between sensitivity and sensibility. Usually, heuristic methods use scoring matrices to calculate the mismatch penalty between two different proteins. In Figure 1, we assigned a unique value for a mismatch (0 in the example), regardless of the parts involved. This works well with nucleotides but not for proteins. For instance, some mismatches are more likely to occur than others and can indicate evolutionary aspects. 17 For this reason, the alignment methods for proteins use score matrices, which associate distinct values with distinct mismatches and reflect the likelihood of a certain change.
High similarity depends on the species being compared and the evolutionary distance between them. For example, identifiable conserved elements between human and macaque are, on average, 90%-95% identical, while in the more distantly related chicken, average identity is in the range of 65%-70%. Here, we are interested to calculate this distance (score), which is highly computationally expensive part of the algorithm.
Proposed Methods
Our method of tiling transformation
The main drawback of the traditional tiling transformation17,18 for loop transformation during parallelization is the limited main memory size for longer DNA sequences and high dependency on the computational resources of the host side. The simple alignment algorithm requires additional storage and communication bandwidth. Therefore, we introduce the new tile-based method, which simplifies the computational model and also handles very long sequences with less data transmission. Our tile-based method can be described as follows: if the matrix can be fully loaded into the memory, do it; otherwise divide the large matrix into tiles to ensure that each tile fits in the processor's memory as a whole and then calculate the tiles one by one or in parallel. Here, we introduce our new method of tile reduction, an OpenMP and OpenMPI tile aware parallelization technique that applies reduction on multidimensional arrays. This new method uses tile reduction implementation including the required OpenMP and OpenMPI API extension and the associated code generation technique for aligning DNA sequences, with the matrix of size 4 × 4 as shown in Figure 2a, where the number of rows (r) and columns (c) are 4 and 4, respectively. In our algorithm, the number of rows will be diagonal, which can be calculated as (r + c - 1), ie, 7 as shown in Figure 2b. Elements of each submatrix correspond to one tile. And for each diagonal row, one file will be created where computed values will be stored and destroyed synchronously. This new parallel implementation is shown in pseudocode Listing 3.1.
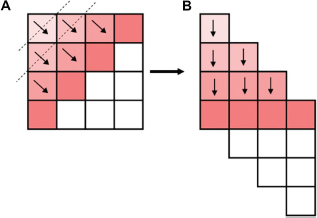
Wave-front structure of sequence alignment algorithm.

Pseudo code corresponding to Figure 2.
The wave-front algorithm is an important method used in various scientific applications. Here, the sequence alignment algorithm also executes each element in wave-front manner, and our new tiling transformation method will also execute in wave-front manner. The computing procedure is similar to a frontier of a wave to fill a matrix, where each block's value in the matrix is calculated based on the values of the previously calculated blocks – left, top, and top-left blocks. This wave-front pattern of computation is within the chunk and also between the chunks.
In Figure 2a, we can see the wave-front structure for parallelizing the algorithm. Since the value of each block in the matrix is dependent on the left, upper, and upper left blocks, as shown in Figure 2a, blocks with the same color are put in the same parallel computing round. Figure 2b shows the data skewing strategy to implement this pattern. This memory structure is useful because blocks in the same parallelizing group (same color) are adjacent to each other. During programming, we have stored the values in the same file for same colored tiles. For one color blocks (one diagonal), one file is created, which is needed for the calculation of next colored blocks (next diagonal) for which another file is created. When third file gets created, first file (first colored block data) is deleted as it is of no use now.
Thus, for every third step, first file gets deleted. This deletion process is synchronous. This method of tiling has been used in all our parallel implementation – OpenMP and MPI. We study these paradigms for various input sequence sizes, various tile sizes, number of threads for OpenMP, number of processes for MPI, and both threads and processes for hybrid model.
Communication model in OpenMP
In our experimental analysis on OpenMP programming model, we observe that as the input sequence size increases, the optimum performance obtained with respect to time and speedup improves with the decreasing number of threads (working cores). To analyze this result, here in this section, we presented a simple method on how to estimate the overhead of communication in OpenMP loops. The reason behind this observation is probably because of the following reason. The architecture used for these results is 12-core Intel Xeon(R) CPU X5650 with 2.67 GHz frequency. The total number of processors is two with each containing six cores. The layout is illustrated in Figure 3. When running tests with tile sizes 8,192 × 8,192 and 16,384 × 16,384, for example, 4 cores, we must use a minimum of

Data are contained in the shaded memory block, lengthiest gone from CPU11 and CPU12 (reproduced and simplified from Ref. 19).
How the data are exactly located on each core is not known, but assume that the tile size matrix is located as depicted in Figure 3, where the shaded memory block contains the data from the master working core. Now, when the algorithm uses 12 OpenMP threads, all the cores will access the memory block in all iterations of the solver. For the cores to retrieve this memory, the cores that are farthest away from the data might have to be idle for several cycles to get the data to be processed. This will happen even if the data are not distributed as in Figure 3, because of the nature of OpenMP.
The total overhead, Ot, for an OpenMP loop on processors executed by a number of threads can be expressed as

where Tcomm is the time required for communication and Tthreads is the overhead of spawning, destroying, and switching between active threads. For a for-loop in OpenMP, a loop of ith iterations is divided into chunk size of k, so that the number of messages sent is

and the size, m, of the messages are
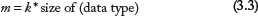
The cost of communication of k bytes is known to be

where Tm is the latency of the respective cache level or memory where the data are located and β is the bandwidth between the processors. Summarizing these formulas, we get a general expression for overhead of communication in an OpenMP for-loop,

There are three variables in this equation that we should look into to provide a better understanding of the estimate, the bandwidth β, the latency Tm, and the chunk size k.
–- The bandwidth could well be related to how many processors share the communication bus, so this variable could be decreasing as the number of processors increases.
–- Tm can be found with the same method as β, only with NULL-size messages. Depending on how many processors participate in the computation, the length of the data bus and how many processors are using the data bus at the same time will influence the latency. As more cores participate in for-loop, it is likely that we end up with a decreasing average latency.
–- The tile size k can be specified statically with the OpenMP keyword schedule (static, tile size). This is however not recommended, since a normal programmer seldom knows the optimal tile size. By not specifying the tile size, it is to be decided in runtime and therefore difficult to know.
Further, the speedup graphs indicate that we get little speedup in our parallel algorithm with this strategy. This could be because of border value exchanges between tiles during the calculation, or that the latency, Tm, is less significant than the bandwidth, β, in Equation 3.5.
If a thread migrates to another CPU for some reason, this migration takes extra time and must be included in the overhead calculation. To test if this was a significant source of overhead, we used the bindprocess() function call to bind the OpenMP threads to a given CPU. However, this did not give any notable performance improvements. Since the bind-process() function did not give any improvement, it might be that threads are already bound to a CPU in IBM's OpenMP implementation or that threads did not migrate before we used the bindprocess() call. The MPI implementation is such that the multiple instances of the same code runs in parallel on various processors. Each processor communicates with the other by message-passing interface and stores its data in its own memory. To distinguish between the processes, a unique id is given to them, which are used to have explicit communication between them. The data are distributed to all working MPI nodes where each individual core will run the code in parallel. The individual cores locate these data in their own memory bank close to it. This avoids the idle time for waiting for memory because of the close proximity to the memory. 20 Here, as all the data are in level 3 cache, MPI processes achieve greater performance. The experimental results based on these observations are discussed in detail in “Experimental results” section. A significant amount of communication overhead is reduced due to sharing of data by several cores on each node. Hence, MPI gives improved performance for large size of files. As for small sequence size, time required for distribution of data to all nodes will be comparatively more.
Experimental Results
The multicore platform that we have used for testing our parallel algorithms for shared and distributed memory structures was as listed below:
64-core Intel 2.53 GHz CPU with 12 MB cache, 12-core Intel 2.83 GHz CPU with 3 GB memory, and 500 GB of global memory, Quad-core Intel 2.83 GHz CPU platform with 3 GB memory, and 500 GB of hard disk, and Intel Core 2 Duo 2 GHz CPU with 2 GB memory.
To evaluate the performance of the sequence alignment algorithm, a representative set of real DNA sequences in FASTA format and BLAST dataset was considered, characterized by a broad range of different lengths (see Table 1).
Input sequences used for experimental analysis.

Comparison of traditional tiling and our tile-based method using OpenMP
Here, the experiments were performed on quad-core Intel 2.83 GHz CPU platform with 3 GB memory, and 500 GB of hard disk. We employ the automatic sequence-generating program to generate different test cases. Hence, different amounts of data sets were used for each result on various architectures.
The general test was executed on four different implementations of the algorithm for DNA sequence alignment. The first one is the serial implementation, the second is the simple wave-front implementation using OpenMP, the third is traditional tile-based implementation, and the fourth is our new tile-based implementation. For long sequences, the simple wave-front version cannot load the entire matrix into the processor memory. Therefore, we select groups of sequences that have variable lengths to test on Linux version. The experimental results are shown in Table 2.
Performance comparison of three different algorithms on Quad processor.

In the serial version, the compiler's optimization methods have a great effect on the manner of accessing memory; however, in multicore processor, this is not true. Because multicore processor has a special hierarchy of memory structure, the access time for different levels of the hierarchy greatly varies. For example, global memory has an access time of about 500 cycles and the on-chip memory such as a register of only one cycle. This is due to the reason that multicore processor does not provide a good mechanism to optimize memory accesses at a compiler level. Also we can see from the results that, among four versions of the algorithm, our tile-based method gives better results.
Here, the experiments were performed on the platform that has a 12-core Intel 2.83 GHz CPU with 3 GB memory, and 500 GB of global memory (hard disk). The selected tile size and the size of the sequence will significantly affect the algorithm's efficiency. In our test, we focus on how the number of threads, which gets created corresponding to the number of cores of the system, affects the final performance. In our tests, we have fixed the size of the tile to be 1,000 × 1,000 and change the sequence length from 16,000 characters up to 2,56,000 characters. The test results are shown in Figure 4, in which we see that with the growth of the average sequence length, the performance increases massively for all the methods. When the average sequence length exceeds 32,000 characters, the time of sequence alignment will increase much faster. This is probably because the main memory size of this 12-core processor is 32 GB. When the size of the sequence exceeds 32,000 characters, access of data from main memory requires more CPU cycles.

Experimental analysis on 12-core processor for variable sequence length.
The parallelization of algorithm with three different methods as shown in Table 3 shows that our new method of tiling gives better performance compared with the other two. This is emphasized in Figure 5, which shows a pairwise comparison between the overall speedups for all the methods of parallel algorithm. The comparison is done for each pair of sequences. Figure 5 shows that the tile-based implementation is an order of magnitude faster than the simple wave-front implementation, because it only needs to calculate a portion of the dynamic programming matrix. In addition, the time growth of the tile-based implementation is lower. As the sequence length increases, the growth of the calculation time needed by our tile-based method is very low.
Performance assessment of three different algorithms on 12-core processor with tile size 1,000 × 1,000.

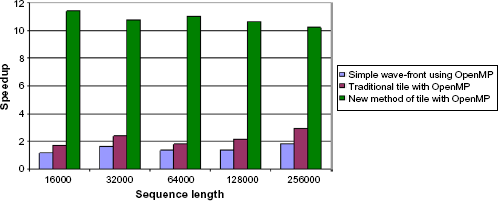
Comparison of speedup for different versions of parallel algorithm on 12-core processor.
Test of tile-based global algorithm on 64-core processor
Here, the experiments are performed on the platform that has a 64-core Intel 2.53 GHz CPU with 12 MB cache. Figure 6 shows the performance evaluation on a 64-core multithreaded processor corresponding to Table 4 for various sequence lengths and computational time in seconds. Here, we also observed that the time required increases from 32,000 character length of the sequence file.

Experimental analysis on 64-core processor for variable sequence length.
Performance assessment of three different algorithms on 64-core processor with tile size 1,000 × 1,000.
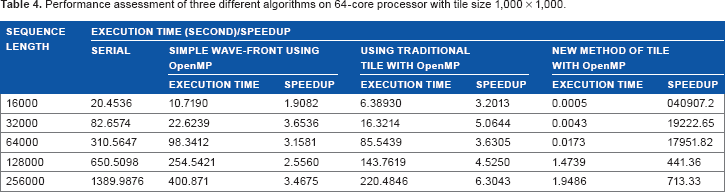
Performance analysis of OpenMP and MPI
The experimental results were performed on (two) Linux-based workstations of Intel Xeon processor with 2.66 GHz of 12 cores each. We have analyzed the execution time and speedup with the parameters as changing number of threads/processes and chunk sizes for OpenMP/MPI, respectively. Figures 7 and 8 show the corresponding execution time and speedup analysis. The results were obtained for OpenMP/MPI implementations where the number of threads/processes generated corresponds to the number of working cores, respectively.
Time analysis for OpenMP implementation on input files of various sizes.

Analysis of speedup for OpenMP implementation on input files of various sizes.
In MPI programming, as each process processes different pieces of same code independently and simultaneously, it gives more speedup on distributed architecture compared to shared memory system. As shared memory uses fork–join model, it stops improvement in speedup after some point, which is mainly due to limited memory.
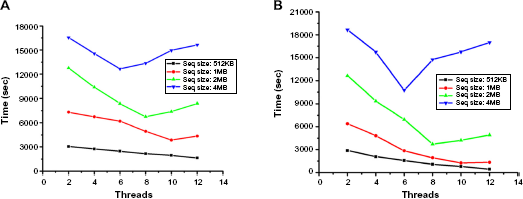
Both the implementations were tested on publicly available GenBank database 20 over various length sequences; four sample input files that have benchmark alignments are taken. The file size used for our experiment consists of 512 KB, 1 MB, 2 MB, and 4 MB base pairs long. Tables 5 and 6 illustrate our analysis on both paradigms, with graphical representations shown in Figures 7, 8 and 9, 10, respectively. The figures show that MPI implementation gives an improved speedup for large sequences compared to OpenMP implementation. We also get adequately acceptable speedup for small and medium sequences. The highlighted entries in Table 5 show that, with the increase in sequence size, the optimum performance was obtained with the decreasing number of threads (working cores) for both tile sizes. For sequence size of 512 KB, 1 MB, 2 MB, and 4 MB, the best performance was obtained on 12, 10, 8, and 6 threads, respectively. Similar analysis can be observed from the curves of graphs of Figures 7 and 8 for time and speedup, respectively. This is because of the communication overhead in OpenMP loops as discussed in “Communication model in OpenMP” section. The highlighted entries in Table 6 show that the optimum performance was obtained with the increasing number of processes and tile size with the corresponding increase in the input data size. That is, for input data size of 512 KB, 1 MB, 2 MB, and 4 MB, the optimum performance is obtained on 12, 16, 20, and 24 processes, respectively. The similar analysis is observed from the graph of Figures 9 and 10 for time and speedup, respectively. This is because communication between the processes reduces as the size of data sent to the slave process increases, which would be otherwise more for small data size. Also with the increase in number of processes, more data get computed simultaneously. Hence, for large input data size, MPI gives improved performance.
OpenMP – performance evaluation on different tile sizes.

MPI – performance evaluation on different tile sizes.


Time analysis for MPI implementation on various sequence size input files.
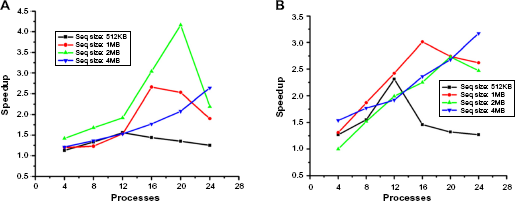
Analysis of speedup for MPI implementation on various sequence size input files.
Conclusion
In this paper, we have
proposed a new method of tiling transformation, which has no limitation on the size of the sequence files as well as tile size. parallelized and optimized existing sequence alignment method using OpenMP and MPI model with our technique for data distribution to handle better memory utilization. for estimating overhead in for-loops, a general communication model is proposed in OpenMP. The detailed study of OpenMP and MPI has been done carefully that concluded that MPI provides the better performance particularly when the size of input files surges.
We are able to efficiently apply our new tile-based nested parallelization with OpenMP to three production codes to increase the scalability on multicore processors. Using OpenMP, the performance increase is nearly linear to the number of processors with minimal modifications to the source code, thus impacting neither the algorithmic structure nor the portability of the code. It is clearly shown that shared memory parallelization is a suitable way to achieve better runtime performance or applications in biological algorithm. The experimental results show that our tile-based parallel algorithm achieves better speedup compared with the serial, simple wave-front and traditional tiling implementation of the global algorithm. For OpenMP loops, we advised a general expression to model the overhead of communication based on number of loop iterations, block size, latency, and bandwidth. Here, we conclude that as the architecture and size of the tile changes, the optimum performance obtained also changes. Using “C” std 99 version, we have also improved the way memory was allocated and freed. Here in this work, we have done the analysis of sequence alignment for two sequences. This study can be further extended by aligning multiple sequences to find the distance between them. Further, these distances can be used to construct the evolutionary tree called phylogenetic construction.
Author Contributions
Conceived and designed the experiments: DDS. Analyzed the data: DDS. Wrote the first draft of the manuscript: DDS. Agree with manuscript results and conclusions: DDS. Jointly developed the structure and arguments for the paper: DDS, SRS. Made critical revisions and approved final version: DDS. Both authors reviewed and approved of the final manuscript.