
Abstract
Bovine rhinitis viruses (BRVs) cause mild respiratory disease of cattle. In this study, a near full-length genome sequence of a virus named RS3X (formerly classified as bovine rhinovirus type 1), isolated from infected cattle from the UK in the 1960s, was obtained and analyzed. Compared to other closely related Aphthoviruses, major differences were detected in the leader protease (Lpro), P1, 2B, and 3A proteins. Phylogenetic analysis revealed that RS3X was a member of the species bovine rhinitis A virus (BRAV). Using different codon-based and branch-site selection models for Aphthoviruses, including BRAV RS3X and foot-and-mouth disease virus, we observed no clear evidence for genomic regions undergoing positive selection. However, within each of the BRV species, multiple sites under positive selection were detected. The results also suggest that the probability (determined by Recombination Detection Program) for recombination events between BRVs and other Aphthoviruses, including foot-and-mouth disease virus was not significant. In contrast, within BRVs, the probability of recombination increases. The data reported here provide genetic information to assist in the identification of diagnostic signatures and research tools for BRAV.
Introduction
Bovine respiratory disease (BRD) is a leading cause of death of feedlot cattle in the USA. Annual costs associated with BRD total more than one billion dollars. Environmental stress, compromised host immunity, and virus infection predispose the animal to bacterial lung infection (broncho-pneumonia). 1 Primary virus infections damage the respiratory tract and subdue the host immune system. The respiratory tract commensal bacteria cause opportunistic secondary infections and lung pneumonia under immune-compromised conditions (caused by viral infection and other factors). In fact, previous studies have reported bovine viral diarrhea virus, bovine respiratory syncytial virus, bovine herpesvirus 1, parainfluenza 3 virus, adenovirus, and bovine coronavirus from cattle with BRD.2–6 Although more than 40 years ago, studies concluded that bovine rhinitis viruses (BRVs) (then known as bovine rhinoviruses) cause mild respiratory disease in cattle,7–10 it is a surprise that BRVs are sparsely studied. This is partly due to the fact that the virus grows poorly in cell culture and is known to cause only self-limiting mild respiratory illness.
Based on the genomic organization and serologic and molecular characteristics, bovine rhinitis A virus (BRAV) and bovine rhinitis B virus (BRBV) along with foot-and-mouth disease virus (FMDV) and equine rhinitis A virus (ERAV) are classified as four species in the
In the new era of deep sequencing, metagenomic profiling of biological samples has yielded a greater diversity of pathogens than was previously detectable. For example, recently, Ng et al. 7 , using metagenomic approach, detected bovine influenza virus, bovine adeno-associated virus, bovine parvovirus, picobirnavirus, and multiple strains of BRAV and BRBV along with earlier reported viruses from BRD cattle samples. In another independent study, Hause et al. 8 showed that BRAV and BRBV commonly circulate in BRD cattle. These findings raise a concern that similar to other BRD-associated viruses, primary infection with BRVs could facilitate bacterial infection. Alternatively, BRVs may themselves cause BRD in some cases.
Previously, we characterized the near full-length genome length sequence of BRBV strain EC11 (EU236594, previously known as bovine rhinovirus type 2) and compared virus-encoded RNA elements and proteins with the related Aphthoviruses.13,14 BRVs can be safely handled in a biosafety level 2 laboratory and have thus received more attention after Osiceanu et al. 15 proposed BRBV as a surrogate model for FMDV anti-viral development, the latter being a virus that requires strict biosafety level 3 conditions for any manipulation. The reason for the strict laboratory guidelines for handling FMDV stems from the fact that it is the etiological agent of an economically devastating disease and is one of the most infectious animal viruses known. Like BRV and other Aphthoviruses, of which FMDV is the prototypic member, FMDV is a +ssRNA virus with a large open reading frame (ORF) flanked by two highly structured 5’ and 3’ untranslated regions (UTRs). As with other Aphthoviruses, the FMDV ORF encodes a polyprotein that is subsequently proteolytically cleaved into a series of intermediate and mature proteins, and often this ORF is subdivided into three regions namely P1, P2, and P3. Just upstream of the P1 region exists the coding sequence for the Lpro, which is preceded by two different AUG start codons that results in two different isoforms of the enzyme (Lab and Lb). The P1 region encodes four structural proteins: VP4 (1A), VP2 (1B), VP3 (1C), and VP1 (1D), while P2 and P3 encode nine nonstructural proteins: 2Apro, 2B, 2C, 3A, 3B1, 3B2, 3B3, 3C protease (3Cpro), and 3D RNA-dependent RNA polymerase (3Dpol).
The Lpro of FMDV cleaves a variety of host factors in the cell cytoplasm, which prime the host cell environment for virus replication and help to subvert the host innate immune response. As such, Lpro is considered as a significant virulence factor. In an effort to produce a useful vaccine platform, “leaderless” FMDV constructs were characterized, showing considerable attenuation relative to parental virus. In a separate study, the Lpro of FMDV was functionally exchanged with BRBV Lpro to develop an FMDV–BRBV chimera vaccine.
16
This vaccine showed promise in controlling FMDV infection under controlled experimental conditions. However, we speculated that the observed similarity between FMDV and BRV has another face to reckon with. Organisms containing similar (homologous) sequences may undergo recombination. In fact, multiple in-depth examinations of publicly available Picornavirus sequences have revealed evidence of significant recombination events among the +ssRNA genomes of
A large percentage of the published
A potential recombination between BRV and FMDV has never been observed in the past, thereby making BRVs an attractive target to explore its possible exploitation in designing chimeric vaccine candidates. Therefore, we sought to address the probability of recombination between the FMDV and BRV genomes and, in particular, BRAV RS3X. To this end, we performed several
This study details new and important genomic information regarding Aphthoviruses in general and specifically, we report a new member of the bovine rhinoviruses, which is BRAV. Several bioinformatics tools employed in this study help to elucidate the molecular diversity of Aphthoviruses, and help to distinguish BRAV from other members of the virus lineage. The knowledge gleaned herein and the applications of these bioinformatics tools will assist other researchers to investigate novel viral pathogens.
Materials and Methods
RNA Isolation, cDNA Synthesis, and Nucleic Acid Sequencing
Viral RNA was extracted from a field sample of BRAV RS3X (Ide and Darbyshire, 1969) using an RNeasy mini kit (Qiagen NV). First strand cDNA synthesis was performed using SuperScript® III First-Strand Synthesis System for reverse transcription-polymerase chain reaction (RT-PCR; Thermo Fisher Scientific) with an oligo-dT primer and approximately 1 μg of total RNA as the template. Different fragments of RS3X cDNA from the 5’ UTR end to the 3’ UTR end were amplified in a model PTC-200 thermal cycler (MJ Research Inc.) and using the Phusion High Fidelity PCR kit (Thermo Fisher Scientific) with specific primer pairs complementary to BRAV. The PCR reaction conditions used were according to the manufacturer's recommendations for amplification products between 5 kilobase (kb) pairs and 9 kb pairs. The 5’ end of the genome between the putative poly(C) and VP3 coding regions was isolated by using a commercially available rapid amplification of cDNA ends (RACE) kit (SMART™ RACE; Clontech Laboratories Inc.). A 3’ anchored cDNA was synthesized using the complimentary gene specific primer (VP3-R1: 5’ TTGGGCCTCACTCAGAGTGGTGGGGGGAT-3’) and by following the manufacturer's protocol. By using the Advantage® 2-PCR enzyme system, 5’ RACE reactions were carried out using anchor (universal primer mix) and gene-specific primers (VP3-R2: 5‘-TGGGTCCGCGGTGATGGGACTAGTGGTGC-3’) according to the manufacturer's recommendations for amplification of products ranging from 1 kb to 5 kb. PCR products were purified using a PCR-purification kit (Qiagen NV), and the integrity of amplicons was confirmed on a 1% agarose gel. Sequences corresponding to the ends of the purified amplicons were obtained by direct sequencing with specific primers designed from previously determined partial BRAV (SD-1) sequence. Subsequent sequence data were determined by using a “primer walking” strategy in which primers for sequencing were designed based on ongoing sequence determination. All sequencing reactions were carried out using the Big Dye Terminator cycle sequencing kit (Thermo Fisher Scientific) and analyzed on a PRISM 3730xl automated DNA sequencer (Thermo Fisher Scientific).
Sequence Assembly and Genome Annotation
Nucleotide sequences were assembled and analyzed with Sequencher (Gene Codes Corporation). The sequence reported in this work has been deposited in the GenBank database under accession number KT948520. All other GenBank accession numbers are indicated either in figures or figure legends.
Analysis of Nucleic Acid and Amino Acid Sequences in Relation to Related Aphthoviruses
Molecular Phylogenetic Analysis by Neighbor-Joining Method
The evolutionary history of BRAV RS3X was inferred by building P1 and 3Dpol coding region phylogeny in
Selection Pressure Analysis
Selection pressure was evaluated by determining the natural selection mechanisms acting on the codons of the ORFs of Aphthoviruses and BRVs. These mechanisms were determined using hypothesis testing using phylogenies package under the Datamonkey web-server (www.datamonkey.org).
30
The d
Recombination Analysis
Possible recombination events among different Aphthoviruses and BRVs were assessed separately using RDP v.4.65. 34 In default mode, RDP, GENECONV, CHIMAERA, MAXCHI, BOOTSCAN, PHYLPRO, LARD, SISCAN, and 3SEQ algorithms were utilized to detect potential recombination events between the input sequences. 34
Structural Modeling
Homology models of BRAV RS3X 2B and FMDV A24 Cruzeiro 2B were built using the hepatitis C virus structure (protein data bank (PDB): 2MTS). VP1 of FMDV A24 Cruzeiro and BRAV RS3X were modeled using the FMDV type O capsid (PDB: 1FOD Chain 1) as a template. All of the homology models were prepared on SWISS-MODEL workspace.35–39 The stereochemical quality of the models was further validated with PROCHECK. 40 Structures were rendered using USCF-Chimera 1.10. 41
Results
Genome and Genome Encoded Proteins
We sequenced the near full-length genome of BRAV RS3X (7,267 nucleotides) from a putative poly(C) tract at the 5’ UTR end to the poly(A) tail at the 3’ end.
5’ UTR
Starting with the 5’ UTR poly(C) of BRAV, RS3X shares 64.12%, 54.00%, and 41.03% nucleotide sequence identity to FMDV (A24 Cruzeiro), BRBV, and ERAV, respectively.
Lpro
ORF scan analysis suggests that Lpro of BRAV RS3X is smaller in comparison with the Lab form observed in other Aphthoviruses including FMDV, BRBV, and BRAV isolates BSRI 4 and 140032-1. As shown in Figure 1, the Lpro active site (highlighted in yellow color)
42
and translation initiation factor-binding sites (highlighted in cyan color) are conserved among BRAVs. Importantly, in BRAV RS3X, these sites are identical to other members of the

(
P1 Region
The structural protein VP4, the first encoded protein from the P1 region, shows the highest conservation among Aphthoviruses as well as BRVs. However, BRVs encode a longer VP4 protein than other Aphthoviruses. The N-terminus of VP2 is remarkably conserved among all Aphthoviruses. The rest of the VP2 protein and the entire VP3 and VP1 of BRVs and ERAV are distinctively different from that of FMDV. Additionally, the characteristic RGD tripeptide, which serves as the cell surface-binding site in the VP1 G-H loop of FMDV,46–48 is lacking in all other Aphthoviruses, suggesting an alternative mechanism of their interaction/attachment to the cell surface (Figs. 1A and B). Considering the absence of the RGD tripeptide in BRAV RS3X VP1, structural models of FMDV A24 Cruzeiro and BRAV RS3X were prepared and compared. Structural models of the two proteins when superimposed on each other show that the G-H loop region is completely disordered in the BRAV RS3X VP1 molecule, which reinforces the speculation that a different virus-receptor interaction occurs on the cell surface (Fig. 2Bi).

(
Non-Structural Proteins
2A containing a C-terminal ribosome-skipping motif NPG↓P encoded by all Aphthoviruses (and many other Picornaviruses) is similar in size. The N-termini of 2A show greater variation, suggesting a lesser functional significance for this region.49,50
The most obvious differences between BRVs (including BRAV RS3X reported here) and FMDV (Figs. 1A and B) are the sizes of the 2B and 3A proteins as well as the number of 3B (VPg) peptides encoded. The N-terminus of the 2B “viroporin”51–53 in all Aphthoviruses, but FMDV bears a significant deletion toward the N-terminus. TMMH (membrane protein topology prediction method) web server analysis revealed that 2B of BRAV RS3X lacks a trans-membrane region that is observed in FMDV A24 Cruzeiro (Figs. 2Ai and ii). However, when we built a homology model of BRBV RS3X 2B using hepatitis C virus (HCV) p7 viroporin (PDB: 2MTS), the viroporin domain appeared to be conserved in BRAV RS3X and was very similar to HCV p7. In fact, the pore lining histidine residue is only present in BRAV RS3X, and FMDV 2B lacks the pore lining α-helix altogether, suggesting a different mechanism for FMDV 2B interaction with intracellular membranes (Fig. 2Bi).
The non-structural protein 3A contains two hydrophobic residues (L38 and 41 in FMDV) that are supposedly important for its attachment to intracellular membranes. Although the hydrophobicity of the amino acid residues corresponding to residues 38 and 41 of FMDV 3A are conserved among all the Aphthoviruses, the smaller 3A of BRAV RS3X and ERAV followed by BRBV indicates a divergence in the membrane association of 3A of these viruses from FMDV. 54 The functional significance of smaller 2B and 3A proteins remains undefined and invites further investigation.
Furthermore, the 2C-ATPase is conserved in BRAV RS3X as well as other Aphthoviruses when compared to FMDV. Importantly, the amino acid residues corresponding to residues 116, 160, and 207 of FMDV 2C that are critical to 2C activity are conserved in BRV RS3X and other Aphthoviruses compared here (Figs. 1A and B).
FMDV is unique among Picornaviruses with respect to encoding three non-identical copies of the 3B (VPg) protein in its P3 region, although possible examples of two copies of VPg are speculated for members of the
Finally, as expected, both the 3Cpro and 3Dpol (highlighted in yellow color) are most conserved and catalytically important residues of the two proteins (highlighted in yellow color) are absolutely conserved in all the viruses compared herein.56,57
Phylogeny
We constructed neighbor-joining phylogenetic trees of the structural protein region P1 as well as non-structural protein 3Dpol, the former being most diverse and latter being most conserved. The BRAV RS3X sequence reported in this study and all of the classified Picornaviruses (54 species in 31 genera, as shown in Fig. 3) were utilized to illustrate the evolutionary space of BRAV RS3X in
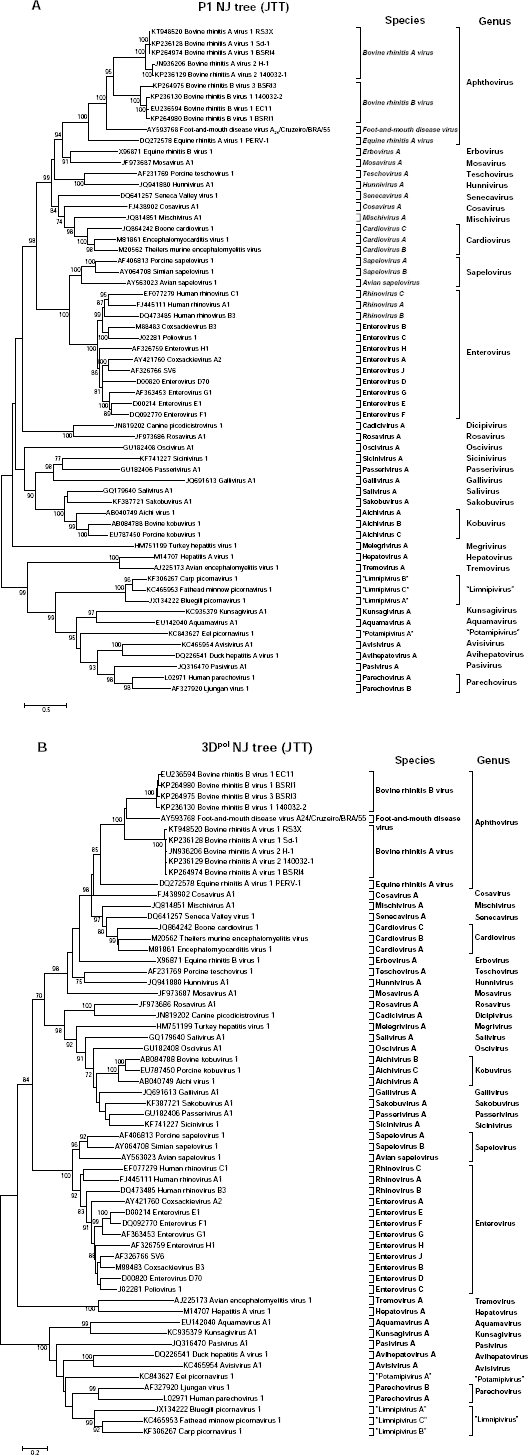
Neighbor-joining phylogeny trees of the P1 structural protein region (
The NJ-JTT phylogeny tree of the 3Dpol region showed similar clustering of Aphthoviruses (Fig. 3B). BRAV RS3X and the isolates BSRI4, SD1, H1, and 140032-1 form a cluster distinct from FMDV A24 Cruzeiro, BRBV, and ERAV. Finally, the order of relatedness exactly matched with that of the P1 region phylogeny.
Selection Pressure Analysis
Positive selection is a major mechanism of RNA virus evolution. Therefore, we first analyzed the selection pressure between Aphthoviruses that are the closest relatives to BRAV by including the BRAV RS3X strain, FMDV A24 Cruzeiro, BRBV, and ERAV by various methods included in the Datamonkey web-server. The results shown in Table 1 show that the SLAC method did not detect any codon site subject to positive selection between these viruses. However, the FEL method detected codon sites 489 and 832 in the structural protein P1 region and codon site 1958 in the 3Dpol region under positive selection pressure. The IFEL method that computes site-wise selection only on the internal branches of the phylogeny tree detected 14 sites subject to positive evolutionary selection, with ten sites in the 3Dpol region and four in the structural protein P1 region. Together, these data suggest that the 3Dpol region has the higher prevalence of evolutionary selection. SLAC and FEL methods detected 363 and 425 sites under negative evolutionary pressure, whereas IFEL unexpectedly found only 3 sites under negative selection. It appears that the selection pressure detected by IFEL, by virtue of working on the internal branches of the phylogeny tree, detects mostly the selection events within species and hence BRAV RS3X and BRBV, being more closely related, would yield such unexpected results.
Distribution of positively or negatively selected sites among Aphthoviruses.

Analysis of selection pressures acting among BRVs by the SLAC method did not detect any positive selection. The FEL method detected one codon site, 680 in the structural protein region under positive selection. However, the IFEL method detected six codon sites subject to positive selection. Four of these sites (614, 617, 680, and 811) are in the structural proteins, one (139) in Lpro, and one (1806) in 3Cpro. All three methods identified numerous sites under negative selection pressure in BRVs with SLAC, FEL, and IFEL detecting 511, 1143, and 843 sites subject to negative selection, respectively. The MEME method, which represents an advancement to the FEL method and detects positive selection sites that would be detected negative in the FEL method, detected 6 codon sites under positive selection among Aphthoviruses: three in the 3Dpol region at positions 2054, 2331, and 2394; one at position 49 in the Lpro region; and sites 735 and 884 in the P1 structural region (Table 1). Among BRVs, the MEME method detected 22 sites subject to positive purifying selection and distributed evenly along the ORF, clearly suggesting the existence of strong positive selection within BRV species (Table 2).
Distribution of positively or negatively selected sites among Bovine rhinitis viruses.

Determination of Potential Recombination Events
During recombination, two molecules of DNA or RNA that carry matching sites (homologous sequences) exchange their segments to yield novel combinations. In fact, recombination is considered to contribute significantly to RNA virus evolution. When analyzing a group of nucleotide sequences for the probability of recombination events, the most common first step is to search for so-called “recombination breakpoints” in the existing sequences. Multiple algorithms have been designed to dissect an alignment of several nucleotide sequences for local regions that exhibit the hallmarks of a recombination breakpoint. The RDP software package uses multiple algorithms, such as RDP, GENECONV, CHIMAERA, MAXCHI, BOOTSCAN, PHYLPRO, LARD, SISCAN, and 3SEQ. In its default mode, RDP calculates even the least possible event. However, a recombination event should only be considered significant if it is evidenced by multiple methods. In this study, we considered an event to be significant only when evidence was provided by four or more methods. In this way, we could take into account the results produced by four out of nine methods employed. Such a strategy has been applied to interpret the recombination events detected using RDP. 58 Table 3 shows the results from an analysis of BRAV RS3X and three other viruses: FMDV A24 Cruzeiro, BRBV, and ERAV. Due to several breaks produced in the ORF due to inclusion of both the 5’ and 3’ UTR, these regions were excluded from the alignment and only single ORFs from the Lpro to 3Dpol regions of these viruses were aligned and included for recombination detection. RDP that recognizes even the remotest possible event, recognized six potential recombination hot spots in the ORF of these viruses, and BRAV RS3X was involved in five of them. However, none of these were confirmed by more than four methods set up as cut-off, and hence we concluded that the possibility of inter-species recombination between different Aphthoviruses analyzed herein is negligible.
Detection of potential recombination events between BRAV RS3X and closely related Aphthoviruses using ORF only.

= The actual breakpoint position is undetermined (it was most likely overprinted by a subsequent recombination event). Minor Parent = Parent contributing the smaller fraction of sequence. Major Parent = Parent contributing the larger fraction of sequence. Unknown = Only one parent and a recombinant need be in the alignment for a recombination event to be detectable. The sequence listed as unknown was used to infer the existence of a missing parental sequence. NS = No significant.
We then analyzed different BRVs for potential recombination events using RDP. As shown in Table 4, in contrast with inter-species recombination being insignificant within species, BRVs carry multiple recombination hot spots distributed throughout the ORF. No less than six of these were reproducible by multiple methods highlighting that these points are real recombination break points.
Detection of potential recombination events bovine rhinitis viruses using ORF only.

= The actual breakpoint position is undetermined (it was most likely overprinted by a subsequent recombination event). Minor Parent = Parent contributing the smaller fraction of sequence. Major Parent = Parent contributing the larger fraction of sequence. Unknown = Only one parent and a recombinant need be in the alignment for a recombination event to be detectable. The sequence listed as unknown was used to infer the existence of a missing parental sequence. NS = No significant.
The findings of these algorithms (Tables 3 and 4) for the assessment of recombination potential are consistent with other published studies regarding the potential of Picornavirus genomes to undergo recombination events. 34 We concluded based on these findings and what was previously described in the existing literature that the likelihood of detecting recombination events between FMDV and BRV could be described as highly remote. We also inferred that given the absence of prior exchanges between the genomes with two different sample pools, the chance of future exchanges would be novel and highly unlikely.
Discussion
In this study, a novel strain RS3X of BRAV was sequenced to its near full-length. Annotation of features and comparison of sequence alignment of BRAV RS3X to related Aphthoviruses and BRVs revealed several unique features in its genome. As one would expect, the BRAV RS3X sequence exhibits conserved features of the BRAV species. Major differences between the prototypic Aphthovirus FMDV and BRAV RS3X were observed in the architecture of capsid and non-structural proteins 2B and 3A. Only the capsid protein VP1 of FMDV displays a cellular integrin-binding arginine-glycine-aspertate (RGD) tri-peptide motif in its G-H loop.46–48 The remaining three Aphthoviruses (BRAV, BRBV, and ERAV) lack this essential cellular receptor-binding site as revealed by structural modeling and analysis of sequence alignment, suggesting a different mechanism of cell attachment for these viruses. In fact, the ERAV crystal structure revealed that it binds the cell surface via sialic acid. 59 The 2B viroporin of BRAV RS3X is significantly smaller than FMDV, and lack of a C-terminal trans-membrane helix of the former indicates a diversion in its topology or function from FMDV. Surprisingly, the modeled structure of the 2B protein from FMDV and BRBV suggest that the latter preserves the membrane pore-lining histidine residue, whereas FMDV 2B lacks this characteristic feature of a viroporin molecule. 60 Conservation of active sites and other functionally critical residues in Lpro, 2Apro, 3Cpro, 3Dpol, and membrane anchorage residue in the 3A protein of BRAV RS3X when compared with Aphthoviruses (FMDV, BRBV, and ERAV) and within BRV species reinforces the functional retention of the molecular biology of Aphthoviruses in BRAV RS3X.
Neighbor-joining phylogenetic trees constructed with the most diverse P1 region and most conserved 3Dpol region parallel each other, confirming the accuracy of phylogeny tree inference. In both analyses, RS3X clustered with BRAV isolates, confirming its classification in that species. The close proximity of BRAV RS3X with isolates BSRI4 and SD-1 suggests a closer evolutionary relationship between these isolates, thus confirming their classification in the BRAV-1 serotype (formerly known as bovine rhino virus type 1) to which it is antigenically related. Isolates H-1 and 140032-1 form a distinct cluster which has been serologically classified as a second serotype, BRAV-2 (formerly known as bovine rhino virus type 3).
Potential recombination breakpoints in the genomes were detected with sites identified flanking the P1 region containing the structural protein-coding sequences and others further downstream in a region containing the non-structural protein-coding sequences. The evaluation of potential recombination breakpoints was performed using distinct sets of algorithms dependent upon which software package was employed. The similarity in results obtained using both algorithmic sets strengthens the final conclusion that recombination events are unlikely between the FMDV and BRV genomes within the regions encoding the structural proteins. Based on examination of the recombination analysis data of Aphthoviruses and BRVs, we conclude that the inter-species recombination events involving FMDV, BRAV, BRBV, and ERAV do not seem likely. However, within each of the two BRV species, the occurrence of multiple recombination breakpoints confirms an underlying phenomenon of homologous recombination-mediated generation of diversity in
Selection pressure analysis is a significant methodology for depicting a common lineage for rapidly evolving organisms, and has been employed extensively in inferring the evolutionary information of viral populations.32,58,68–71 Given that multiple analytical methods were employed in this study, there is a high degree of confidence added to the interpretation that positive selection is not operating between BRV and other Aphthoviruses. The observed higher number of positive selection breakpoints among BRVs is not a surprise and reinforces their common ancestry. As expected, negligible evidence of single-point positive selection sites proves a parallel but distinct lineage of the different Aphthoviruses included in this study.
In conclusion, the data from this study provide valuable information on Aphthoviruses and, more precisely, BRAV to serve as the genetic basis for future studies. Detailed knowledge of the evolution and divergence of Aphthoviruses at the molecular level could aid in the design of BRV-based molecular diagnostic tools and new bio-therapeutics.
Author Contributions
Generated and analyzed the data: DR, PL, SJP, MEP, NJK, ER. Wrote the first draft of the manuscript: DR, PL. Contributed to the writing of the manuscript: DR, PL, ER, NJK. Agreed with manuscript results and conclusions: All authors. Jointly developed the structure and arguments for the paper: DR, PL, ER. Made critical revisions and approved final version: DR, PL, NJK, ER. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgment
We thank Elizabeth Schafer and Anna Kloc for their critical reading and valuable comments.