
Abstract
Bacterial autotransporters represent a diverse family of proteins that autonomously translocate across the inner membrane of Gram-negative bacteria via the Sec complex and across the outer bacterial membrane. They often possess exceptionally long N-terminal signal sequences. We analyzed 90 long signal sequences of bacterial autotransporters and members of the two-partner secretion pathway in silico and describe common domain organization found in 79 of these sequences. The domains are in agreement with previously published experimental data. Our algorithmic approach allows for the systematic identification of functionally different domains in long signal sequences.
Keywords
Introduction
Bacterial autotransporters translocate via the Sec complex across the inner membrane of Gram-negative bacteria and translocate themselves across the outer membrane.1,2 This is accomplished by a translocator domain at the C-terminus of the autotransporter which adopts a β-barrel fold within the outer membrane 3 resembling a porin-like domain. 1 The trimeric autotransporter consist of an N-terminal signal sequence, a central “passenger domain”, and a β-barrel forming translocation unit. 3 The β-barrel domain is necessary for the secretion of the passenger domain and connected via an α-helical linker region. 4 Bacterial autotransporters have been found in many Gram-negative bacteria and are often associated with virulence factors such as adhesion, biofilm formation, aggregation, invasion, and toxicity. 5 For translocation across the inner bacterial membrane autotransporters possess N-terminal signal sequences. 2 In 2007 Dautin and Bernstein reported around 10% of the know auto-transporters to contain a signal sequence with more than 50 residues. These N-terminal signal sequences exhibit a tripartite organization (n, h, c) as described by von Heijne. 6 According to this nomenclature, “n” refers to an N-terminal region of the signal peptide which varies in length and often contains charged residues. The “h” or core region is a hydrophobic stretch required for the interaction between the signal peptide and SRP. 7 “c” refers to the signal peptidase cleavage site. Additionally they can be roughly divided into two domains: i) an N-terminal extension of about 25 residues, ii) a C-terminal part that resembles a signal peptide. 3 This division is in two domains, where one is like a functional signal peptide and is strikingly similar to the “NtraC model” which has recently been introduced by the writers as a general model of long eukaryotic signal peptides. 8
Henderson et al reported at least 80 proteobacterial autotransporters with a signal sequence of at least 40 residues and published a list containing 46 sequences. 1 The authors propose four different regions based on hydrophobic and charged residue distribution (N1,H1,N2,H2) and a C region (cleavage site) following the standard n, h, c organization of export signals according to von Heijne. 6 Desvaux et al continued this approach and termed the N2 and H2 region the “extended signal peptide region” (ESPR). 9 They propose that the ESPR may be important for additional functions besides targeting. In this report, we extend and formalize this approach by proposing a dual domain organization proposed by our algorithm.
Materials and Methods
We analyzed 16 autotransporters and two-partner secretion sequences published by Szabady et al 10 and 35 further long signal sequences of bacterial autotransporters taken from Henderson et al. 1 Two-partner secreted proteins are known to possess an N-terminal conserved region important for their secretion. 11 Additionally we performed a sequence database search in UniProtKB/SwissProt Release 14.7 12 using the sequence retrieval system (SRS, Release 7.1.3). 13 We searched for proteobacterial sequences with an annotated similarity to an autotransporter domain and a signal sequence of at least 40 residues, resulting in 56 sequences. Of those 56 sequences 39 were not considered in the work of Henderson et al 1 and Szabady et al. 10 From the sequences considered suitable by the work of Henderson et al 1 Szabady et al 10 and our own database search we assembled a dataset of 90 sequences. The signal peptidase cleavage sites were used as suggested in Henderson et al 1 Szabady et al 10 and for the 39 sequences retrieved via SRS as annotated in SwissProt UniProtKB/SwissProt Release 14.7, respectively. The SwissProt database entries contain sequences with predicted or putative signal sequences.
The following sequences were omitted from our analysis due to minor sequence aberrations between the publications and the UniProtKB database entry: O32591, Q47692, Q54151 and Q8VSL2.
The following to YP_001161762 orthologous sequences were omitted since they possess an identical signal sequence: YP_001719317, YP_001874066, Q1C309, Q1CMJ2 and Q665P5. When two database entries contained the identical sequences one entry was omitted and both accession numbers are given.
In total, 90 signal sequences encompassing more than 40 residues from bacterial autotransporters were analyzed in this study and in regards to their possible internal domain organization.
The 28 long signal sequences not associated with autotransporters were retrivied from the UniProtKB/SwissProt Release 14.7 12 using the Sequence Retrieval System (SRS, Release 7.1.3). 13 We searched for “non-potential” bacterial signal sequences with evidence at protein level and a length of at least 40 residues. All retrived sequences contain the twin-arginine (TAT) 14 signal which leads to export to the periplasm or extracellular space (Suppl. Table 1).
The 228 short bacterial signal peptides associated with autotransporters were retrieved from the UniProtKB/SwissProt Release 14.7 12 using SRS (Release 7.1.3). 13 We searched for proteobacterial signal sequences with less than 40 residues that contain annotated similarities to known autotransporters (Suppl. Table 2).
The detection of the domains was performed using the NtraC algorithm, 8 an algorithmic approach to identifying domains in long eukaryotic signal peptides based on secondary structure aspects. The NtraC model proposes one domain to be essential and sufficient for targeting while rendering the other domain free for additional functions. Here, “N” and “C” denote two potential domains: an N-terminal “N-domain” and a C-terminal “C-domain” predicted by the algorithm. The transition area between both domains is refered to as “tra”. The algorithm works on the complete signal peptide sequence and suggests the domain positions. The N- and C-domains contain targeting signals that are not detectable when the whole signal sequence is regarded as an entity as performed by current prediction software. Until recently six predicted domains have already been tested experimentally in vitro, from which five exhibit the predicted targeting function 8 (Resch and Hiss in preparation).
Results and Discussion
We analyzed 90 long signal sequences of bacterial autotransporters and the two-partner secretion pathway in regards to their potential two-domain (NtraC) organization.
Of the 16 signal sequences collected in Szabady et al 10 14 are predicted to have a two-domain organization (Table 1). Of the 46 autotransporter signal sequences collected in Henderson et al 1 35 are not listed in Szabady et al. Of those 35 sequences 32 are predicted to have a two-domain organization (Table 1). Of the 39 sequences we found via SRS and which are not described in Henderson et al 10 or Szabady et al 2005, 31 are predicted to have a two-domain organization.
In total, from 90 long signal sequences considered in this study 77 (86%) are predicted, by our algorithm, to be organized in two domains.
For two additional sequences (Q2J0N4, CAR56027) an NtraC organization is predicted which in the context of this work could be regarded as a false-positive: No C-domain with a targeting capacity was detected. For Q2J0N4 an N-terminal mTP is predicted by TargetP15,16 and for CAR56027 a signal anchor by SignalP. 17 If these two sequences are included a total of 79 of 90 (88%) signal sequences are predicted to be organized in two domains.
The two-domain organization proposed by the algorithm is in agreement with the ESPR of Desvaux et al 9 and the conservation of the “N-terminal extension” reported by Szabady et al 10 within a margin of ±5 residues.
Szabaday et al 10 further reported a conserved sequence pattern in the N-terminal extension of autotransporter and two-partner secretion systems signal peptides. 10 This conservation is also present in 43 of 46 sequences compiled by Henderson et al. 1 For the long signals sequences extracted via SRS, the conserved pattern is only present in three sequences (Table 1, no. 52–54). One of these three (no. 54, Q8CWC7) is a Pic variant of a different E.coli strain (Table 1, Nr. 4). The remaining 36 sequences found via SRS do not show the conserved sequence motif reported by Szabady et al 10 although they are annotated to contain a domain similar to autotransporters. This might argue for the sequences of Szabady et al 10 to form a group. Nevertheless, we found that the SRS sequences have the same domain structure in their long signal peptide as the sequences reported by Szabady et al 10 and Henderson et al. 1 The N-terminal region of this group of autotransporters may have a different function not requiring this conserved motif pattern.
We want to highlight the case of the long signal peptide of EspP. EspP is an extracellular serine protease of E. coli which is divided into four subtypes α, β, γ and δ of which α and γ are proteolytically active.18,19 The long signal peptide of subtype EspPα contains the conserved sequence pattern reported by Szabady et al 10 and for which experimental results were published by Peterson et al. 20 These authors showed that residues 23–55 can act as an independent targeting signal. In 2006 Peterson proposed the N-terminal extension of the signal sequences to mediate an interaction with an unknown cytosolic factor or to induce an unusual signal peptide conformation prior to protein translocation. 21 Notably, the analysis of the 55 residue signal sequence of EspP by our algorithm identified a two-domain (NtraC) organization:
NtraC analysis of 90 long bacterial signal peptides.

Nr. 1–16 taken from Szabady et al. 10 ; Nr. 17–51 taken from Henderson et al 10 ; Nr. 52–90 retrieved via SRS.
gives the position of the last residue of the signal peptides as used in the publications or annotated in UniProtKB v. 14.7.
NtraC organization of the sequence.
Length of the predicted N-or C-domain. Numbers in brackets refer to alternative possibilities for truncation of the domains. A targeting abbreviation in brackets means: only that length combination leads to the targeting function in brackets.
/+: gram negative or gram positive signal peptide.
strain = Sakai.
strain = E2348/69.
strain = EB1.
SP: eukaryotic signal peptide, SA: signal anchor gram.
strain = 042.
strain = K–12.
strain = CFT073.
strain = EH41.
strain = 4797/97.
mitochondrial targeting peptide.
strain = Sakai.
strain = 042.
strain = K–12.
strain = H10407.
strain = ML308–225.
strain = APEC13.
strain = O78:H11.
strain = O6:H1.
strain = K12.
N-Domain (residues 1–26): unknown function,
C-domain (residues 27–55): predicted secretion signal for Gram-negative bacteria.
The algorithm thereby proposed the same functional domain Peterson et al described experimentally.
We would like to stress that the NtraC algorithm is based on sequence information only and not influenced by the existing proposed fragmentation of long signal peptides. Our prediction method is therefore unbiased for the analysis of new sequences.
A further surprising result is the prediction of mitochondrial targeting peptides (mTP) for the proposed N-domains of the long bacterial signal peptides. In 17 of 90 (19%) cases the N-domain of a bacterial signal sequence is predicted as mTP (Table 1). Short bacterial signal peptides associated with autotransporters are in 29 of 228 (13%) cases predicted as mTP. As the presence of arginine is a typical feature for mTPs15,16 this could, in our case, lead to a prediction of a sequence as mTP if arginine residues are abundant. The positive charged residues are thought to form amphiphilic α-helices. 22 This high abundance of positive charges (Table 2) has also been observed in the extended N-region of bacterial autotransporter signal sequences by Peterson et al. 21 They reported a high net positive charge to be common in the N-terminus of serine protease autotransporters.
The automatic assignment “mTP” should thus not to be regarded as a perfect functional prediction but as the detection of a feature, namely the high abundance of charged residues. In 1994 Izard and Kendall reported that although a positive charge in the N-terminus may not be absolutely required for secretion 23 a net negative charge or zero charge could result in considerably decreased rates of export.24–26 While Dierstein and Wickner reported that the N-terminal regions is not strictly required for processing by signal peptidase, 27 Peterson et al demonstrated that the positive charges in the N-terminal part of the bacterial signal sequences may influence SRP recognition. 20
To investigate the role of charged residues in silico in the context of long signal sequences of autotransporters and their potential domain organization we counted the occurrence of charged residues in the N- and C-domain of all 79 autotransporter sequences predicted to be two-domain organized (Table 2). The border between the N-domain and the C-domain (transition area, “tra”) often contains charged residues. To take this into account the border between both domains was alternating, and included (+tra) or excluded (–tra) from the domains for the calculation (Table 2).
If the border between the domains was regarded as part of the C-domain(+tra), positively charged residues (His, Lys, Arg) occur approximately 1.6 times more often in the N-domain compared to the C-domain. Negatively charged residues (Asp, Glu) occur 2.3 times more often in the N-domain(–tra) compared to the C-domain(+tra). This difference becomes even more prominent if the border between both domains is counted as part of the N-domain(+tra) leading to 2.8 times higher occurrence of positively charged residues and 4.2 times higher occurrence of negatively charged residues in the N-domain compared to the C-domain.
Mean occurrence of charged residues given in one letter code in 79 NtraC-organized autotransporter signal sequences the length of the transition area (tra) is up to eight residues.

Domains of the NtraC algorithm. 8
Excluding or including the transition area (domain border) predicted by the NtraC model.
Standard deviation in brackets.
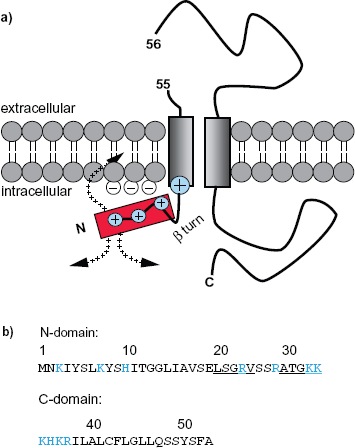
Cartoon of a possible membrane orientation of the signal peptide of EspP.
This charge bias is an argument that charged residues may represent an inherent difference between the N- and the C-domain. The nearly threefold increase in deviance between the N-(+tra) and C-domain(–tra) indicates that not only the presence of charged residues is of importance but also their position, favoring the N-terminal domain or between the two domains. The relative position curtly before the targeting signal in the C-domain could represent a characteristic feature.
The observed abundance of charged residues in the N-domain was also reported for the long signal peptides from vertebrata analyzed by us previously.8,28 The authors therefore propose that a potential additional function of the N-domain in long signal peptides is related to the abundance of positively charged residues in Gram-negative bacteria as well as in vertebrata. This is in agreement with the observation made by Peterson et al20,21 regarding a high net positive charge of the N-terminal part of the signal sequence and its potential influence on SRP recruitment. The NtraC algorithmic approach can be used to check individual observations, and pinpoint the sequence part that might be relevant for such an SRP interaction.
A further hint towards a mechanistic aspect arises from to the secondary structure aspect of the NtraC model. As the C-domain of the signal peptide with its hydrophobic core is embedded in the membrane or the Sec complex during translocation, the N-domain may be kept in a defined angle to the membrane due to a predicted β-turn in the border between the N- and C-domain. The positive net charged of the N-domain could have the effect of keeping it outside and on top of the membrane. This might provide the means for the recruitment of other proteins (Fig. 1). We further report priliminary in silico results that 43 out of the 90 (48%) long signal peptide sequences of long autransporters and 21 out of 28 (75%) long bacterial signal peptides not associated with autotransporters could form an amphipathic helix. We compared this to short bacterial signal peptides associated with autotransporters and found that 34 out of 228 (15%) could form an amphipathic helix. The requirement to form an amphipathic helix was to possess nine adjacent amino acids in a helix in a window of 18 residues. In a second approach we allowed the adjacent nine e.g. polar residues to be interrupted by one e.g. non-polar residue and vice versa. Now 68 out of 90 (76%), 25 out of 28 (89%) and 58 out of 228 (25%) could form an amphipathic helix (Fig. 2). While one must keep in mind that short sequences in general provide less amino acids to form an amphipatic helix at all, we still report a tendancy of long singal peptides to form alpha helices.
Conclusion
We present an extensive analysis of 90 long bacterial autotransporter signal sequences predicting in 86% of the sequences, a common two-domain organization. The described organization is in agreement with published experimental data and allows the identification of potential new domains in silico in long signal sequences. We corroborate the importance of charged residues in bacterial signal sequences and emphasize their position near the N-terminus as possible regularity. The approach highlights the relevance of charged residues in long signal sequences.

Multiple sequence alignment of 79 NtraC-organized autotransporter signal sequences (Matlab R2009a, Bioinformatics Toolbox Version 3.3).
28 long signal sequences not associated with autotransporters.

228 short bacterial signal peptides associated with autransporters.

Footnotes
Acknowledgement
We are most grateful to Dr. H. Bernstein for critical and constructive comments on the manuscript.
The authors report no conflicts of interest.