
Abstract
Bovine respiratory disease (BRD) is the most common economically important disease affecting cattle. For developing accurate diagnostics that can predict disease susceptibility/resistance and stratification, it is necessary to identify the molecular mechanisms that underlie BRD. To study the complex interactions among the bovine host and the multitude of viral and bacterial pathogens, as well as the environmental factors associated with BRD etiology, genome-scale high-throughput functional genomics methods such as microarrays, RNA-seq, and proteomics are helpful. In this review, we summarize the progress made in our understanding of BRD using functional genomics approaches. We also discuss some of the available bioinformatics resources for analyzing high-throughput data, in the context of biological pathways and molecular interactions. Although resources for studying host response to infection are available, the corresponding information is lacking for majority of BRD pathogens, impeding progress in identifying diagnostic signatures for BRD using functional genomics approaches.
Keywords
Introduction
Despite research for decades and investments made in genomics to understand disease progression, bovine respiratory disease (BRD) continues to cause considerable economic losses for the cattle industry. BRD has a complex etiology that involves a multitude of interactions among the host, various pathogens, and the environment. Studying host–pathogen interaction (HPI) networks in BRD has the potential for identifying diagnostic markers or signatures that are indicative of disease and its severity. Functional genomics approaches (often referred to as “omics” approaches) generate genome-scale expression data for mRNA, protein, metabolites, and non-coding RNA, and applying these methods to BRD research can expedite the development of diagnostics. The list of genes/proteins identified by high-throughput approaches require analysis in a systems biology framework, ie, in the context of protein–protein interaction (PPI) networks, to decipher the underlying regulatory mechanisms that are responsible for a given phenotype or biological response. Studying gene/protein expression in a systems biology framework requires a comprehensive description of all parts of the system (structural annotation), identifying the interactions among the parts (protein–protein, protein–DNA interactions, etc), identifying regulatory mechanisms that govern these interactions, and finally developing descriptive mathematical models and iterative refinement of these mathematical models to generate predictive models of the system. At present, applying this entire workflow is not feasible for bovine–pathogen interactions that result in BRD. However, functional genomics approaches allow the generation of testable hypotheses from high-throughput data using gene ontology (GO) enrichment, pathways, and network analyses. Regardless of the data analysis strategy, bioinformatics tools, databases, and algorithms are required for leveraging functional genomics data for BRD diagnostics. The proposed functional genomics-based workflow is described in Figure 1.
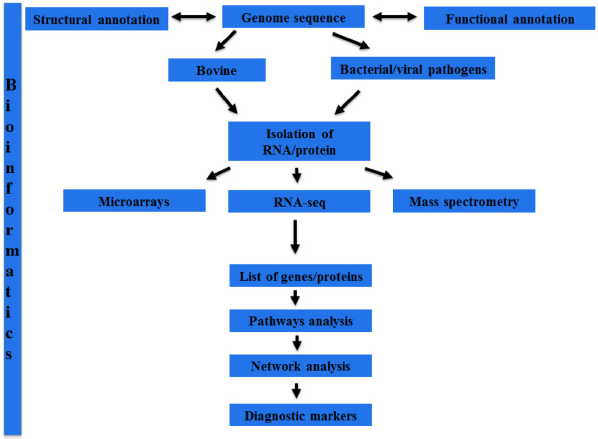
A flowchart describing functional genomics-based approach for BRD diagnostics. The availability of sequenced genomes for bovine and BRD pathogens enables the study of genome expression using various high-throughput approaches such as microarrays, RNA-seq, and proteomics, which often generate a list of genes/proteins. Continuous updates to existing structural and functional annotations enhance the quality of functional genomics findings. At the core of these annotation approaches as well as analysis of functional genomics data in the context of biological pathways and networks is the necessary bioinformatics analysis and resources. An integrated systems biology framework for analyzing functional genomics data is expected to identify accurate biomarkers for prediction of BRD resistance and susceptibility, as well as disease stratification.
For the purposes of this review, we used the following definition of functional genomics: “Functional genomics uses genomic data to study gene and protein expression and function on a global scale (genome-wide or system-wide), focusing on gene transcription, translation and protein-protein interactions, and often involving high-throughput methods” (http://www.nature.com/subjects/functionalgenomics). Therefore, genome-wide association studies that identify genetic variations responsible for BRD susceptibility/resistance to aid beef cattle breeding programs and studies that focus on the role of small non-coding RNAs in posttranscriptional gene regulation are not discussed in this article. The reader is pointed to reviews in literature that focus on microarray and RNA-sequencing (RNA-seq)1–7 and proteomics8–11 methodologies and challenges associated with the data analysis.
In this review, we describe BRD, enumerate challenges associated with BRD diagnostics, and outline the bioinformatics methods/resources that enhance the structural and functional annotation of BRD genomes for analyzing gene/protein expression data for hypothesis generation. Progress made toward BRD diagnostics using proteomics, microarrays, and RNA-seq is summarized.
Bovine Respiratory Disease
BRD is the most common and economically important disease affecting cattle. 12 BRD causes 70%-80% morbidity and 40-50% mortality in feedlot cattle. 13 BRD is temporally associated with factors such as weaning, transportation, and nutritional and social alteration. 14 These environmental factors, often referred to generically as stress, are host dependent, highly interconnected, largely immeasurable, and consequently poorly understood.15–17 The most common viruses involved in the BRD complex include bovine herpesvirus-1 (BHV-1), bovine parainfluenza virus type-3 (BPIV3), bovine respiratory syncytial virus (BRSV), and bovine viral diarrhea virus (BVDV). 18 Typically, these viruses infect the upper respiratory tract, resulting in rhinitis, tracheitis, and bronchitis, and generally do not cause major lung damage but do predispose the lower airway to bacterial invasion. 19 The primary bacterial agents, often obtained from BRD mortalities and associated with severe inflammation and lung damage, include Mannheimia haemolytica, Pasteurella multocida, Histophilus somni, and Mycoplasma bovis. 20 The bacterial agents are commensals, found commonly in the respiratory tract or on other mucous membrane surfaces21–23 and cause lung damage only when host defenses are diminished because of increased stress and/or concurrent viral infection.24,25 Nearly all (99.8%) cattle identified with clinical BRD receive antimicrobial treatment, and antimicrobials are used extensively in disease prevention. 26 In addition to the cost incurred in the production phase, evidence clearly indicates that BRD has negative effects on carcass weight, marbling, and other carcass value factors.27,28 Animals affected with BRD do not always exhibit clinical symptoms.29–31 Animal health products and management strategies to prevent infection and reduce the negative effects of BRDC pathogens over the past 50 years are yet to be conclusively linked to increased disease control. 32 Thus, BRD diagnostics continues to be a problem to date.
Existing Measures for BRD Diagnosis
There are very limited reliable tools to identify animals affected with BRD.32,33 In vivo sampling techniques, including brochoalveolar lavage, transtracheal wash, and nasopharyngeal swab, have been described and used primarily to procure samples for agent identification.34–36 Clinical signs are used to determine disease onset and severity, but clinical signs are subjective, difficult to standardize, and nonspecific for BRD.27,31,37 However, observation of clinical signs remains the predominant tool for detection of BRD in vivo. Generally, cattle that become ill have moderate risk of developing pulmonary pathology and exhibiting lung lesions at harvest, but the majority of cattle with evidence of pulmonary pathology at harvest are not detected as being ill in life.27,29,31,38,39 Infrared thermography 40 and ultrasonography 41 are often used for diagnosis, along with lung biopsy, 42 continually reporting rumen temperature, 43 blood and breath biomarkers,44–46 haptoglobin measurement,30,47,48 and eating and drinking behaviors. 38 However, none of these approaches has shown promise for in vivo field use. As such there is no “gold standard” for BRD diagnostics as noted in a recent review of laboratory tests for BRD. 49
Thus, progress in BRD research is at an impasse and continued investigations focusing on independent elements (ie, pathogen or host) are unlikely to result in major breakthroughs in BRD research. This situation has been well recognized, and research focusing on identification of markers associated with genetic resistance to BRD has been initiated through the Bovine Respiratory Disease Coordinated Agricultural Project (BRD CAP), 50 which will among other data generate gene expression profiles for single-agent challenge using the majority of BRD pathogens. 51 For field application, diagnostic criteria that allow early disease identification and assessment of lung damage would allow treatment strategies to be tailored to the condition and help identify animals with poor predicted outcomes, similar to the way procalcitonin has been advocated for use in human bacterial lung infections. 52 Such diagnostic criteria would enable more rational treatment decisions, limit antimicrobial use to those most likely to respond, and decrease suffering by identification of animals unlikely to survive.
Functional Genomics for Studying BRD
The complex interplay of bacteria, viruses, and environmental factors that results in BRD is poorly understood and is unlikely to be fully elucidated using reductionist strategies. Pathogens have evolved numerous strategies to successfully invade their hosts (acquire nutrients and evade immune system), and host defense systems are continually evolving to combat infections.53,54 The availability of genome sequences for bovine55,56 and BRD pathogens, including BHV-1,57–59 BVDV, 60 BRSV, 61 BPIV3, 62 M. haemolytica,63–67 P. multocida subsp. multocida str. Anand1_cattle (contigs: GCA_000291645.1), H. somni, 68 and M. bovis,69,70 facilitates transcriptomics and proteomics studies to identify molecular functions, pathways, and networks that are involved in BRD pathogenesis. To detect, manage, and treat BRD, it is critical to elucidate the key molecules in the host–pathogen interplay and generate systems biology models for emergent behavior. Systems biology focuses on the systematic study of complex interactions in/between biological systems and aims to produce quantitative models that are predictive. 71 A number of biological processes, including inflammation, apoptosis, and thrombosis, are involved in the pathogenesis of BRD. Identification of these processes by the “omics” approaches, with subsequent evaluation in a systems biology framework, and linking the findings back to traditional end points will help develop diagnostic tools specific to BRD.
Improving Genome Sequence Quality: Structural Annotation
High-throughput technologies, such as microarrays, RNA-seq, and proteomics, provide readout of gene/protein expression on a genome scale. Studying the gene/protein expression in cattle or BRD pathogens could identify disease mechanisms for developing sound diagnostics. Involvement of genes/proteins in BRD pathology can only be determined provided they are annotated, ie, demarcated in the genomes. The process of identifying and defining all the functional elements within a genome sequence is defined as “structural annotation”. Structural annotation is the foundation for functional genomics approaches that focus on measuring the expression of all known genes/proteins in an organism. Genome structural annotation is concurrent with genome sequencing efforts. This initial annotation is based on in silico predictions that rely on sequence similarity. Although computational approaches have high accuracy of prediction, using experimental data to update existing in silico predictions 72 is warranted. Experimental annotation at the RNA level using tiling arrays and single-nucleotide resolution RNA-seq results is expected to accurately identify transcriptional start sites, operon structures (in bacteria), exon–intron boundaries, and novel non-coding functional elements such as small RNAs (sRNAs) and long non-coding RNAs (lncRNAs). Of relevance to this review article, open reading frames with protein-coding potential that were missed in the initial annotation can be identified by experimental genome annotation methods. We reported RNA-seq based expression profiling studies for two bacterial BRD pathogens, H. somni 73 and M. haemolytica. 74 In H. somni strain 2336, we identified 94 sRNAs, of which 82 sRNAs were not reported elsewhere. We also identified 38 novel potential protein-coding open reading frames and 278 operon structures in the genome. Compared to a nonvirulent strain 129Pt, we found that ~30% of the identified sRNAs were unique to the virulent strain 2336, indicating that a number of the newly identified sRNAs in strain 2336 may be involved in strain-specific adaptations. Our RNA-seq based transcriptome map of M. haemolytica resulted in the identification of 14 novel protein-coding regions and 44 potential novel sRNAs. While both of these studies reported the identification of novel non-coding RNAs and protein-coding regions, confirmation of these findings by additional experimental approaches is necessary before this new knowledge can be incorporated into existing databases for functional genomics studies. Although the role of non-coding RNAs in regulating gene expression is well known, it still remains poorly studied in BRD. Using computational pipelines and nonprotein coding transcripts from expressed sequence tags (ESTs), thousands of non-coding RNAs (ncRNAs) were identified in the bovine genome. 75 Inspection of the genomic distribution of the ncRNAs, majority of which were not reported previously, indicated their transcription adjacent to open reading frames with a possible involvement of ncRNA in cis-regulatory roles. These computational predictions for the existence of ncRNAs in bovine systems need confirmation by complementary experimental annotation approaches. The proposed whole-genome sequencing in the 1,000 Bull Genomes Consortium Project is expected to provide annotated sequence variants and genotypes of key ancestor bulls, which will enhance the quality of the genome enormously. Sequencing results of 234 76 bulls has facilitated the mapping of monogenic as well as complex traits in cattle. Analysis of functional genomics data in the context of such information-rich bovine genome sequences will revolutionize our understanding of BRD for developing diagnostics.
Enhancing Genome Sequence Utility: Functional Annotation
Analysis of functional genomics data often generates a list of genes/proteins that are differentially expressed in response to a biological perturbation (biotic or abiotic). For understanding the mechanisms of BRD pathophysiology, it is important that all genes/proteins identified as being associated with BRD have functional information, ie, biological function. Bioontologies are controlled vocabularies for describing biological concepts such as gene functions, and the relationships that exist between concepts, to facilitate data sharing and computational analysis of high-throughput data sets.77–79 GO is one of the most commonly used bioontologies,80–82 which describes gene product functions. GO is the prevailing standard for functional annotation and consists of three separate ontologies: Biological Process, Cellular Component, and Molecular Function. BRD researchers can access GO for bovine gene products at AgBase. 83 AgBase is a curated, open-source, Web-accessible resource for carrying out functional analysis of agricultural plant and animal gene products. AgBase provides GO for bovine gene products and also has tools for functional genomics data analysis. AgBase tools like GORetriever fetch available GO for an input gene list and GOslimviewer summarizes GO annotations at a higher level. For bovine gene products that lack GO, functional domains with GO annotations in other species are identified by GOAnna by a Basic Local Alignment Search Tool (BLAST) search, enabling the transfer of GO annotations from other species to bovine gene products, when appropriate. Recognizing that not all gene products have information that can be annotated to GO consortium standards, AgBase captures community annotations to provide information for any gene product.
Bioinformatics Resources for BRD Research
In the following sections, we summarize some of the bioinformatics resources available for analyzing functional genomics data in the bovine host and BRD pathogens. Although not comprehensive, this section describes some of the fundamental resources for conducting “omics”-based BRD research. Web links are provided for these resources in Table 1 for easy access.
Bioinformatics resources available for analyzing BRD functional genomics data.

The Bovine Genome Database
A genome sequence is not very useful as a linear string of ATCGs unless there is a way to visualize the sequence. The architecture of genome browsers such as GBrowse 84 is a combination of database and query-based visualization interface that allows users to view the genome. This interface allows the users to add metadata, which are displayed as a separate track that can be turned on or off. The bovine genome database 85 is a database that was developed to facilitate the visualization and annotation of the bovine genome sequence. This is a valuable resource that allows the bovine research community to utilize the existing two competing versions of genome assembly for research. As summarized by the bovine genome database, “the Btau_4.6.1 assembly includes the Y chromosome, while UMD3.1 version does not include the Y chromosome; however, bovine research community perceives UMD3.1 assembly as the more contiguous version”. This database displays both bovine genome assemblies in a GBrowse format and includes information regarding the official gene set, gene models from data repositories such as RefSeq and Ensembl, non-coding RNAs, repeats, pseudogenes, single-nucleotide polymorphisms, markers, quantitative trait loci (QTLs), and alignments to complementary DNAs, ESTs, and protein homologs. Because the bovine genome database is connected to the Bovine QTL viewer, it facilitates the identification of candidate genes underlying a quantitative trait loci.
AgBase 83 hosts GBrowse for three BRD bacterial pathogens, namely, M. haemolytica strain PHL213, H. somni strain 2336, and P. multocida strain 3480. The purpose of these microbial genome browsers is to primarily display proteogenomics data for structural annotation. The genome itself could be viewed independent of additional data such as peptide-level data to gain insights into the organization of functional elements in these bacterial genomes. The University of California Santa Cruz genome browser 86 hosts H. somni 129Pt and M. succiniproducens, strains that do not cause BRD but can be used for comparative genomics.
Bovine Gene Atlas
A comprehensive description of the abundance of transcripts in all tissues is often referred to as a gene atlas, and availability of this information can help identify tissue-specific expression. The bovine gene atlas 87 is available through the AgBase website. This resource was developed by applying next-generation sequencing to determine the RNA abundance in 92 adult, juvenile, and fetal cattle tissues and 3 cattle cell lines. The results are displayed in a GBrowse format, allowing the end user to navigate and access the project results. Bovine expression atlas results confirmed the expression of 16,517 annotated protein-coding loci in the genome. Analysis of BRD functional genomics data in the context of lung tissue expression atlas will help identify lung-specific gene/protein expression during BRD infection, when compared to expression in the nonrelevant tissues for disease onset and progression.
Resources for Pathways Analysis
Analysis of “omics” data sets for drawing biological conclusions often requires linking a gene/protein set to pathways. The HUGO Gene Nomenclature Committee Comparison of Orthology Predictions 88 search tool helps identify bovine–human orthologs to tap into available functional annotation of human genes that can be transferred to bovine genes. Pathway resources for BRD pathogens are available through databases such as the Kyoto Encyclopedia of Genes and Genomes (KEGG), 89 Metacyc,90,91 and Unipathway, 92 which have dedicated pages for the individual genomes also in addition to cataloging the annotated pathways. UniPathway is expected to be integrated with detailed UniProt 93 information. Uniprot is a curated database resource for protein functions and pathways. Reactome94,95 is another good source of pathways information. For a comprehensive list of bioinformatics resources relevant to BRD, we suggest the use of Pathguide, 96 a resource that lists all available PPI databases, as well as pathways and networks resources.
InnateDB
While it is important to interrogate the bovine and BRD pathogen genome sequences to identify the genomic context of functional genomics data, it is critical to identify a subset of host genes that constitute the immune response to infection with different BRD pathogens. Innate immune host responses are the first line of defense against pathogens and, when dysregulated, can cause damage to host tissues. To develop a diagnostic marker for BRD that is specific to a particular pathogen, it is important to characterize bovine innate immune responses to all BRD pathogens, in order to determine singe-agent specificities. InnateDB 97 is a repository of >196,000 curated human, mouse, and bovine molecular interactions. There are 3,000 pathway annotations of relevance to cellular systems, including immune-relevant pathways. There are 46 bovine genes with documented roles in innate immune responses in this database. As more information regarding innate immune responses in bovine physiology becomes available from large-scale projects such as BRD CAP, results can be incorporated into InnateDB, which has evolved into a resource that can support systems biology framework for analyzing functional genomics data. Molecular interactions in InnateDB contain contextual information that meets the standards set forth by the minimum information required for reporting a molecular interaction experiment (MIMIx) standards. 98 Because this information includes the evidence for each interaction, the tissue or cell type that the interaction was identified, and other related data, in conjunction with tissue expression atlases, InnateDB will be a powerful tool for identifying tissue-specific immune response networks for BRD diagnostics.
AnimalTFDB
To identify gene regulatory networks in BRD, it is important to analyze functional genomics data in the context of transcription factors (TFs) that are known to be involved in the regulation of gene transcription. TFs have different DNA-binding domains, which form the basis for their classification into different families, and up to 5% of genes in vertebrate genomes are predicted to be TFs. 99 The animal TF database AnimalTFDB has information regarding bovine TFs, cofactors, and chromatin-remodeling factors. 99 A recent update of the database includes evaluation of the expression of TFs from tissues using RNA-seq based expression data. This update to the database also includes a TF prediction server and allows for comparison of TFs from different species for the identification and prediction of this important class of proteins involved in the regulation of gene expression.
HPIDB
The study of HPIs to identify drug targets and develop vaccine strategies has a strong precedent in biomedical research. In infectious disease research, it is important to understand the commonalities between diseases for efficient development of therapeutics. Computational methods such as multitask learning are being applied to test this “commonality hypothesis” for multiple bacterial species, including Bacillus anthracis, Francisella tularensis, Yersinia pestis, and Salmonella typhi. 100 With a multitude of pathogens being associated with BRD, identifying such a commonality in bovine response to pathogens will help provide specificity for BRD diagnostics. To study functional genomics data in the context of bovine–BRD pathogen interaction networks, availability of such HPI data is a prerequisite. Although a number of interaction databases for HPI exist, they are primarily focused on human pathogens. One such resource, VirHostNet,101,102 is a database of curated virus–host molecular interaction data. At present, VirHostNet has only seven HPIs for BVDV and three HPIs for BHV, with very limited number of HPIs. We developed host-pathogen interaction database (HPIDB), 103 which focuses on integrating available HPIs from multiple databases and thus generates a set of nonredundant HPIs for multiple hosts and pathogens. At present, HPIDB contains 23,735 unique protein interactions between 68 host and 567 pathogen species. HPIDB provides data in a format compatible with Cytoscape 104 for visualization of the interaction network. Similar to VirHostNet, HPIDB also allows the transfer of homologous HPIs to species of interest by conducting BLAST searches. Of the total species represented in the HPIDB, at least 20% are bacterial species, while 70% are viruses. Therefore, by using HPIDB, it is possible to get a first-pass computationally predicted HPI network for bovine BRD pathogens, which will help prioritize genes/proteins identified by functional genomics for BRD diagnostics.
In summary, bioinformatics resources available for bovine systems allow rapid analysis of functional genomics data for biological interpretation. Given a list of bovine genes, it is possible to retrieve the available functional GO information using the AgBase 83 tool GORetriever and summarize the GO using GOSlimviewer. For a set of bovine genes that lack functional annotation, GOAnna at AgBase can add GOs based on sequence homology. Pathways resources such as Reactome,94,95 KEGG, 89 Unipathways, 92 InnateDB, 97 TFDB, 99 and HPIDB 103 can each add additional bits of relevant information to the list of genes, such as pathways represented, involvement in host innate immune responses, activity as TFs, etc. Resources such as the bovine genome database facilitate the analysis of the genomic context of bovine genes, while tissue-specific expression patterns can be inferred from the bovine gene atlas. Utilizing resources such as HPIDB 103 allows the visualization of bovine–pathogen interactions inferred from homology as networks in Cytoscape. 104
Proteomics in BRD
Although not comprehensive, a few of the reported proteomics studies in BRD are summarized below. Bronchoalveolar lavage fluid (BALF), obtained by washing the epithelial lining of lungs, is often used to analyze proteins that are representative of pulmonary diseases. 105 BALF is often used to understand respiratory disease mechanisms and to identify novel markers of pathogenicity. 105 Characterization of the changes in protein expression in calves challenged with M. haemolytica by nano-liquid chromatography-tandem mass spectrometry (LC-MS/MS) 42 identified ITIH4, an acute phase protein commonly associated with chronic obstructive pulmonary disease in humans, to be elevated, along with haptoglobin, a known acute phase marker for BRD. Interestingly, increase in the expression of the antimicrobial peptides (AMPs) cathelicidin-1 and −4 were also reported in this study. AMPs, also called as natural antibiotics or host defense peptides, are cationic peptides of the host innate immune response and are considered to be ideal candidates for novel peptide-based therapeutics.42,106 Efforts to catalog bovine AMPs based on prediction tools and databases such as AMPer 107 are under way. A recent study used AMPer to search and identify novel AMPs in the bovine genome and sequence tags from the National Center for Biotechnology Information's Database of Expressed Sequence Tags (NCBI dbEST) project. This study identified 27 AMPs with high confidence and 68 AMPs were predicted from the EST data. 108
A major etiological factor of BRD is stress associated with transportation,32,109 weaning, commingling, 110 preconditioning, sanitation, and air quality, 111 which increases the susceptibility of cattle to bacterial pneumonia. In BRD, stress often correlates with immunodepression, and cattle succumb to bacterial infection by M. haemolytica.44,112 Although studies have identified stress as an etiological agent for BRD, molecular markers and mechanisms associated with stress response at the gene and protein levels remain elusive. Stress-associated changes in BALF were assessed by two-dimensional (2D) gel electrophoresis and mass spectroscopy by comparing nonstressed and stressed calves. 113 In this study, expression of annexins A1 and A5, odorant-binding protein (OBP), isocitrate dehydrogenase, fibrinogen, heme-binding protein, alpha-2-Heremans-Schmid-glycoprotein (a-2-HSglycoprotein), alpha-1-antichymotrypsin, and albumin were significantly altered in response to stress. Similar studies were also performed with BALF to identify the effects of glucocorticoids in BRD by administering dexamethasone to calves. 114 Increased levels of glucocorticoids suppress the immune system, rendering calves susceptible to bacterial and viral infections. This study identified several acute phase markers whose expression was elevated, such as alpha-1-acid glycoprotein (orosomucoid) and alpha-1-antitrypsin. Additionally, the amount of a-2-HSglycoprotein (fetuin) was reduced in BALF. Dexamethasone also increased the expression of two hydrophobic ligand-binding proteins, adipocyte-fatty acid binding protein, OBP, alpha-enolase, cofilin-1, and immunoglobulin J chain. Together, these studies evaluated the changes in the protein signature in response to stress associated with BRD,113,114 underpinning the probable mechanistic pathways. Consistent with these reports, Difference Gel Electrophoresis and MS analysis of BALF showed increased expression of annexins A1 and A2 in calves weaned and transported to the rearing site. 115 Differentially expressed proteins in this study included three antioxidants such as dihydrodiol dehydrogenase-3, peroxiredoxin I, superoxide dismutase, calcyphosin, a calcium binding protein, and a macrophage capping protein. 115 The authors recommend the use of annexin A1 as a potential stress-associated biomarker for BRD.
M. haemolytica can produce membrane vesicles that are made of outer membrane proteins (OMPs). Fifty-eight OMPs from M. haemolytica vesicles were identified by LC-MS/MS 116 and these are potential targets for vaccine development. 115 Cattle vaccinated with OMPs showed remarkably lower lung lesion scores compared to nonvaccinated calves. The identification of these novel OMPs was possible because of the availability of the M. haemolytica genome sequence.65,66 These studies demonstrate the importance of proteomics in identifying novel candidates for developing diagnostics and therapeutics that target either the host or the pathogen.
Proteomics of cytopathic (cp) BVDV identified bovine proteins involved in the immune function of professional antigen-presenting cells during BVDV infection.117,118 Proteins of significant interest included proteins involved in cell adhesion, apoptosis, antigen uptake, processing, and presentation such as the transporter associated with antigen processing, acute phase response proteins (fetuins, serum amyloid A, and apolipoprotein A-II), and major histocompatibility complex class I- and II-related proteins. Proteins involved in professional antigen presentation were significantly altered after BVDV infection. This analysis provided mechanistic insights into BVDV infection and showed that cp BVDV, while promoting monocyte activation and differentiation, inhibits antigen presentation to immunocompetent T-cells and subsequent infection. 117 Follow-up proteomic studies using 2D-LC-electrospray ionization (ESI)-MS/MS characterized the changes in protein expression in bovine monocytes by both cp and noncytopathic (ncp) BVDV infections 119 and revealed signaling pathways that were differentially regulated in cp and ncp BVDV infections. Macropinocytosis signaling, virus entry via the endocytic pathway, integrin signaling, and primary immunodeficiency signaling were activated by ncp BVDV-infected monocytes, while pathways of actin cytoskeleton signaling, RhoA signaling, clathrin-mediated endocytosis signaling, and interferon signaling were identified only in cp BVDV-infected cells. Among the common pathways involved in cp and ncp BVDV infection, acute phase response signaling was the most significant for both BVDV biotypes, while integrin alpha 2b (ITGA2B) and integrin beta 3 (ITGB3) were downregulated by ncp BVDV and upregulated by cp BVDV infection. 119 A proteomics based study reported the characterization of differentially regulated protein kinases in response to cp and ncp BVDV infection in bovine monocytes. 120 Using differential detergent fractionation and 2D-LC-ESI-MS, 378 proteins with homology to known protein kinases or related proteins were identified. This approach found upregulation of three kinases that mediate cellular signaling in response to BVDV, such as urokinase-type plasminogen activator receptor (u-PAR), myristoilated alanine-rich C kinase substrate (MARCKS), and nucleoside diphosphate kinase B (NDPKB). Rho-associated protein kinase 1 (ROK1) GTPase involved in cell differentiation and hexokinase type 3 (HK3), a kinase involved in glucose utilization, were downregulated in bovine monocytes in response to BVDV infection. This provides a molecular basis for sugar starvation in BVDV pathogenesis. The expression of the receptor of activated C kinase, pyridoxal kinase, diacylglycerol kinase, and Bruton's tyrosine kinase was reduced in monocytes infected with cp BVDV compared to those with ncp BVDV, suggesting a role for these proteins in the differential pathogenicity of cp and ncp BVDV viruses.
Transcriptomics in BRD
The past decade has seen several technologies designed to analyze gene expression changes in an organism, a tissue, and individual cells. Techniques to profile gene expression include microarrays, RNA-seq based on next-generation sequencing, custom quantitative reverse transcription polymerase chain reaction arrays, and multiplex branched DNA assays. These techniques have enabled profiling of the gene expression of both the host and the pathogen, as well as identifying critical players that are differentially expressed under different pathogenic conditions associated with BRD. A few transcriptomics studies in BRD are summarized below.
Fatal cases of BRD are because of viral–bacterial synergy and are often associated with BHV-1 and M. haemolytica infections in the lower respiratory tract. 121 Approximately 44,000 transcripts were evaluated in bovine bronchial epithelial cell (BBEC) following exposure to BHV-1 alone, M. haemolytica alone, or both BHV-1 and M. haemolytica. 121 This analysis revealed that M. haemolytica exposure led to a significant increase in the production of inflammatory molecules such as chemokine(C-X-Cmotif)ligand2(CXCL2),interleukin(IL)-6, IL-1α, E-selectin, and IL-8, compared to animals challenged with BHV-1 alone in BBEC. However, in the coinfection model, differential regulation of several markers of inflammation, such as tumor necrosis factor-α, IL-8, toll-like receptor-2 (TLR2), IL-1, CXCL2, and colony stimulating factor 2 (CSF-2), were observed along with vascular endothelial growth factor (VEGF), endothelin 2 (EDN2), intercellular adhesion molecule 1 (ICAM1), and IL-16. Taken together, gene expression profiling of BBE cells by microarrays revealed that the viral bacterial synergy could compound the pathogenesis of BRD. Although follow-up studies confirming these observations are warranted, transcriptomics has identified pathways that go awry during the pathogenesis of BRD.
In another study, gene expression profiling of M. haemolytica A1strain was conducted 6 days postchallenge in pneumonic lungs, 122 after establishment of infection. The in vivo gene expression signature was compared to an in vitro-cultured sample. Forty-four genes were differentially expressed by more than eight-fold. Interestingly, 17 genes were upregulated in vivo, while 27 genes were downregulated. The genes that were downregulated included virulence-associated genes encoding leukotoxin, a capsule biosynthetic enzyme, and a serotype-specific antigen, Ssa, suggesting that expression of these genes is not critical once infection is established. 122
To understand the mechanism by which M. haemolytica adapts to low-iron environments, changes in gene expression under iron-limiting growth conditions were assessed at 15 minutes, 30 minutes, and 60 minutes following iron deprivation. 123 Upregulation of several ATP-binding cassette transporters and receptors was found under iron-limiting conditions, suggesting new mechanisms by which M. haemolytica can compensate for lack of iron. Although limited in number, these studies clearly demonstrate the potential for knowledge discovery for BRD diagnostics utilizing transcriptomics.
The BRD CAP team analyzed pathogen-specific host response in the bronchial lymph nodes of steers recently. 51 In brief, single-pathogen challenges were performed in steers for three different viral pathogens, such as BHV-1, BRSV, and BVDV, and three bacterial agents, M. haemolytica, P. multocida, and M. bovis. Transcriptome profiling by RNA-seq identified differential expressions that were common and unique to each challenge agent. Among the differentially expressed genes, a cumulative total of 5,757 genes were identified as unique to single challenges, among which 200 genes that showed high variance were common to all pathogen challenges. Apart from identifying 25 common genes that were differentially expressed in all the infections, the study reported unique genes that were specific to individual challenges. Differentially expressed genes specific to BRSV infection included FRZB, MT1E, NPTX1, TSPAN18, and CA4, while ALB, ITIH2, KRT24, MMP7, and TTR were unique to BVDV. Infection with M. haemolytica resulted in the differential expression of BMPR1B, COL6A6, and KIR3DL2. EBD and TSPAN1 were differentially regulated in response to M. bovis. Not surprisingly, the panel of genes common to all pathogens mostly comprised genes of the host innate immune response, such as MMP9, S100A8, and S100A9. S100A8 and S100A9 are expressed in neutrophils and monocytes 124 and are known danger-associated molecular patterns that bind pattern recognition receptors in response to inflammation. 125 This study exemplifies the value of functional genomics to decipher pathogen-specific response in host tissue, enabling the development of specific gene panels that could be evaluated for developing BRD.
Concluding Remarks
Current therapeutics and accurate diagnostics for human diseases is possible as the human genome is sequenced and well annotated, enabling the study of gene functions and disease mechanisms, making subsequent development of diagnostics and drug targets 126 easier to accomplish. Unlike human diseases, most veterinary diseases are challenged with lack of complete genomic information and the availability of species-specific research tools. 127 This is true for bovine respiratory disease research aimed at developing diagnostics tools. Although there is some information regarding the genes/proteins in the bovine genome that are implicated in BRD, the information is not comprehensive. Availability of annotated bovine–pathogen interspecies interaction is very rudimentary. On the pathogen side of the equation, knowledge pertaining to intraspecies interactions is lacking or minimal. The ongoing human and mouse ENCyclopedia Of DNA Elements projects126,128 highlight the importance of identifying functional elements in the genome at high resolution. However, such an effort is marginal in veterinary diseases. Although parallels can be drawn from human diseases, the differences in causative agents and interspecies differences has often resulted in failure to identify similar disease mechanisms 127 in veterinary clinical science. Ongoing international effort to conduct whole-genome sequencing of 1,000 bulls 76 will address the deficit in the availability of a high-resolution genome sequence that is well annotated on the host side. The recent formation of an organization of an international scale, the Functional Annotation of Animal Genomes (FAANG) project, aims to generate comprehensive maps of functional elements in the genomes of domesticated animal species. 129 There is a large deficit in our knowledge of BRD mostly because of lack of species-specific gene sequences from the host and pathogen, as well as lack of species-specific immunological reagents to perform molecular analyses. The FAANG project should help bridge this gap in veterinary research and generate central repositories of data, tools, and reagents. In multifactorial diseases such as BRD, high-throughput omics approaches such as transcriptomics and proteomics offer a convenient alternative than the traditional hypothesis-driven research to explore disease complexity. These technologies help compare and contrast disease states, tissue-specific biomarkers, and the micro and macroenvironments in pathogenesis that could affect diagnosis, prognosis, and therapy. In human diseases, considerable progress has been made in the identification of biomarkers using functional genomics technologies with the integration of bioinformatics and systems biology. 130 To maximize the impact of future BRD research, particularly when targeting naturally occurring disease in a field environment, a diagnostic strategy with high sensitivity, specificity, and reproducibility across time and geographical sites is essential. Continued investment in deciphering molecular mechanisms of BRD by utilizing functional genomics, with concomitant investments in improving the genomics infrastructure, ie, bovine and BRD pathogen genome sequences and bioinformatics, and computational biology resources is mandatory for making progress toward developing such BRD diagnostics.
Author Contributions
Wrote the first draft of the manuscript: ANR, BN. Contributed to the final manuscript: ANR, WBE, BN. Jointly developed the structure and arguments for the paper: ANR, BN. Made critical revisions and approved the final version: ANR, WBE, BN. All the authors reviewed and approved the final manuscript.
Footnotes
Acknowledgment
We thank Leslie A. Shack for assistance with this article.