
Abstract
A fast and reproducible protocol was established for enzymatic characterization of plant sesquiterpene synthases that can incorporate radioactivity in their products. The method utilizes the 96-well format in conjunction with cluster tubes and enables processing of >200 samples a day. Along with reduced reagent usage, it allows further reduction in the use of radioactive isotopes and flammable organic solvents. The sesquiterpene synthases previously characterized were expressed in yeast, and the plant-derived Thapsia garganica kunzeaol synthase TgTPS2 was tested in this method. KM for TgTPS2 was found to be 0.55 μM; the turnover number, kcat, was found to be 0.29 s−1, kcat for TgTPS2 is in agreement with that of terpene synthases of other plants, and kcat/KM was found to be 0.53 s−1 μM−1 for TgTPS2. The kinetic parameters were in agreement with previously published data.
Introduction
The determination of enzymatic parameters for newly discovered enzymes is based on analytical chemistry techniques that are often termed as bioanalytical technology. These technologies are used to benchmark and assess enzyme efficiency under different conditions, leading to established values such as KM, kmax, kcat, and kcat/KM, which are used to compare enzymes. One of the first studies on sesquiterpene synthases was performed on partly purified bacterial enzymes some 30 years ago. 1 However, the number of biochemically characterized plant sesquiterpene synthases is still limited, considering the number of available enzymes. This is mainly due to cumbersome procedures with low throughput when obtaining kinetic data.
The compounds produced by plants have recently received considerable renewed attention due to their medical properties and potential as useful compounds. 2 5 Synthetic biology is a rapidly growing field that utilizes biosynthetic pathways to develop “compound factories” that are optimized in possible ways for maximal production.6,7 Modulating bio-synthetic pathways to produce valuable compounds is often complex due to varying functional classes and turnover numbers of implicated enzymes, and as such, no chain is stronger than the weakest link. 8 Every step of the biosynthesis must be optimized to yield a financially viable synthetic production platform. Thus, the understanding of the enzymatic activities is crucial for this to be successful. For production of terpenoids, yeast has been preferred for many years and protocols have been developed to study the complexity, but without purification of the enzymes. 9 11 Recent kinetic studies on plant sesquiterpene synthases have isolated the enzymes by heterologous expression in Escherichia coli followed by purification, whereas yeast has not been used for this purpose. To our knowledge, this is the first report exemplifying the applicability of yeast as a heterologous platform for in vitro assays using purified enzymes.
A typical setup for performing enzyme characterization, which is used in a wide range of literature, employs glass vials with screw lids, where handling seems excessively cumbersome. 12 14 O'Maille et al 13 used automatic sampling to process only single vials sequentially in the analysis phase on GC-MS apparatus, which still relies on a high degree of manual handling of each sample in the actual assay phase as is also needed for the radioactive assay.
High-throughput 96-well plate-based methods using malachite green (MG) or NADPH-dependent fructose 6-phosphate have recently been published.15,16 Both methods avoid the use of radioactive substrates and organic solvents, leading to less waste. The MG- and NADPH-dependent assays are coupled enzyme assays in which the release of PPi (diphosphate ion) is measured. This is fundamentally different from the GC-MS and radiochemical assays where the sesquiterpene synthase product is measured; thus, the MG-and NADPH-dependent assays are indirect kinetic measurements and the controls should include a study of breakdown and turnover of PPi. The drawback of the MG assay is the use of the very toxic compound MG, and the NADPH assay uses β-mercaptoethanol. Both compounds are in general cytotoxic 17 and should be used with extreme care, as with organic solvents and radioactive substrates.
Both the MG- and NADPH-dependent assays utilize 96-well microtiter plates to reduce the handling significantly as also described in the present work. Here, we present a 96-well microtiter plate method based on the use of radioactive isotopes and organic solvents to partition the enzyme products. The present assay also utilizes cluster tubes that can be accommodated in specialized 96-well racks, which are about three times taller than regular microtiter plates. Vardakou et al 15 used regular microtiter plates directly for the assay, which are not applicable for the present study that uses organic solvents.
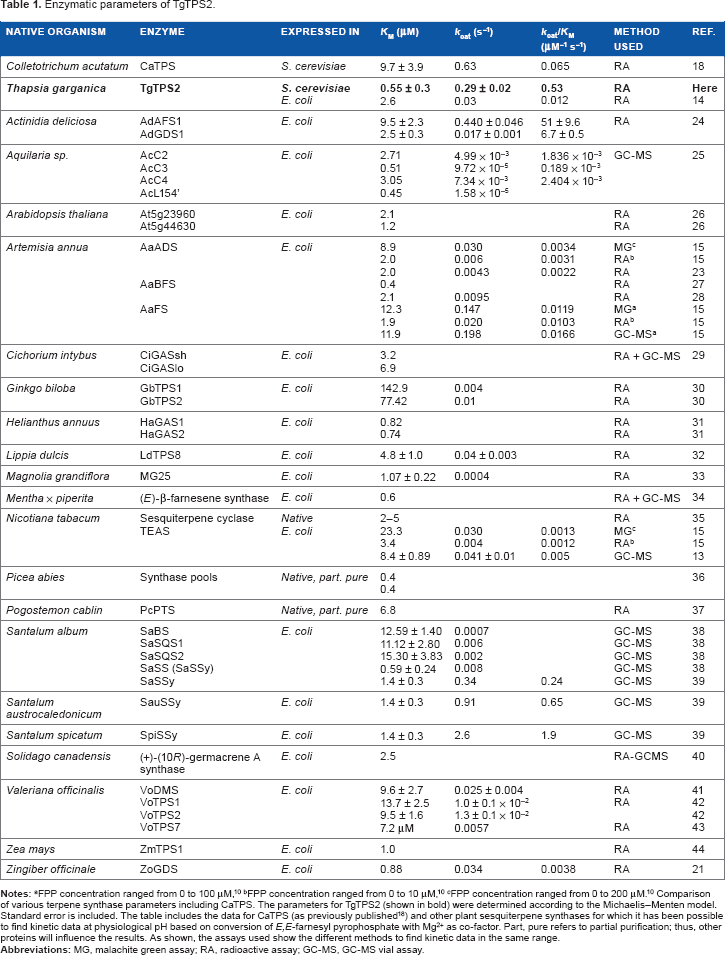
The previously characterized plant sesquiterpene synthase Thapsia garganica kunzeaol synthase TgTPS2 (accession no. JQ290345) 14 was successfully HIS-tagged and expressed in yeast, purified and utilized for developing this method. Yeast was chosen due to an apparently high level of expression and subsequent purification in experiments that followed previous work. 14 Figure 1 illustrates the general strategy employed in the present work. The kinetic data of CaTPS (Colletotrichum acutatum β-caryophyllene synthase, accession no.: KP398851) were previously published utilizing this method and are included in Table 1 as reference. 18
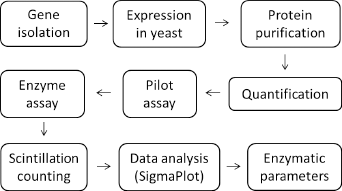
Flow diagram illustrating the present strategy. The strategy used in this study started with gene isolation. The time frame of gene isolation (including transfer to pESO-Leu2d plasmid) may vary and is generally a time-consuming procedure. expression in yeast may take up to a week. Protein purification: 1-2 days; quantification: 1-2 hours; pilot assay: 1-2 hours; enzyme assay: 1-2 hours; scintillation counting: 1 hour; data analysis: 1-2 hours.
Enzymatic parameters of TgTPS2.
Materials and Methods
Materials
The yeast shuttle vector pESC-Leu2d was kindly provided by Ro. 14 BCA reagents/kit was obtained from Pierce and hexane from Fluka. Cluster tubes were purchased from Corning (Product #4412) and nonlabeled farnesyl pyrophosphate (FPP) was from Sigma-Aldrich. 3H-labeled FPP and microplates for scintillation counting were obtained from PerkinElmer. The microplate reader was a MicroBeta 1450 (PerkinElmer). Scintillation liquid was an Ecoscint A from National Diagnostics.
Cloning and expression of sesquiterpene synthases in yeast
Vector pESC-Leu2d [a pESC-LEU2 (Agilent) derivative] was chosen for overexpression in yeast due to the successful prior art concerning artemisinin production.9,10 pESC-Leu2d contains a truncated transcription factor that results in high transcription levels in an unregulated manner.
An N-terminal 6xHIS-tagged version of TgTPS2 was constructed using primers forward-TgTPS2BamHI-HIS6 (GAGAGGATCCATGCACCATCACCATCACCACGCTGTGTATGTTAACTCTACAACA) and reverse-TgT PS2-Xho I (CACACTCGAGTTATGCTGGAATGGGATTTATGAGAAC) from cDNA of T. garganica. 14
The resulting vector was sequenced for validation and then transformed into Saccharomyces cerevisiae wild-type BY4742 and grown overnight (O/N) in 2 × YPD. Transformation was performed as previously published. 19 Transformants were selected on SC-Leu with 2% glucose (SC-Leu-GLU) plates and grown for 4 days, thereafter stored at 4°C. All yeast cultures were centrifuged at 4°C and cultivated at 28°C at 120 RPM in baffled Erlenmeyer flasks of at least 20 mL, keeping 4/5 headspace volume.
For expression of each construct, one large transformed colony was selected and grown O/N in 20 mL SC-Leu-GLU. This starter culture was transferred to 400 mL SC-Leu-GLU and grown O/N. The medium was exchanged with 2 L SC-Leu-GLU, transferred to an appropriate flask, and grown O/N. Finally, the medium was exchanged with 2 L SC-Leu-GAL expression medium (2% galactose instead of glucose) and grown for five to seven hours. One liter culture was harvested, and the pellet was stored at -80°C until further processing. The remaining culture was allowed to grow for further 8-12 hours before harvesting. The two time points (5-7 hours and 15-17 hours) were expected to represent a useful time window for assessing the optimal expression. A total of 15-17 hours appeared to be optimal for both enzymes used in this study.
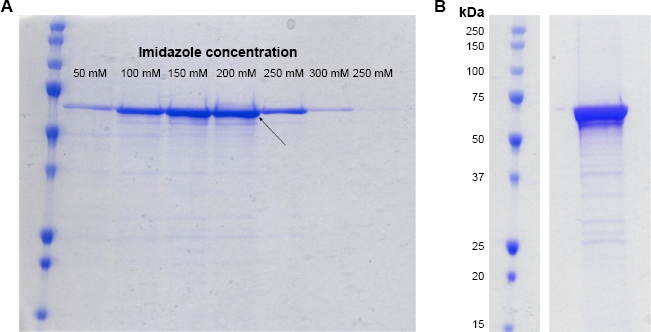
For purification, the pellet was thawed on ice and resuspended in 50 mL binding buffer (BB: 50 mM Tris-HCl pH 7.5, 25 mM imidazole pH 7.5, 500 mM NaCl, 10% glycerol, 1 mM PMSF). The cell suspension was lysed on a Constant Systems Tissue Lyser at 38 kPSI. A total of 30 mL rinse using BB was pooled with the first fraction. The lysate was centrifuged at 4000 RCF for 30 minutes and followed by 18,000 RCF for 1 hour. The supernatant was filtered through a 0.45 μm syringe filter and incubated with Nickel NTA Agarose resin (Qiagen) on a rotator at 4°C for two hours or O/N. The resin was washed with five column volumes BB, and then eluted with imidazole gradient elution buffer (EB 1-8: 50 mM Tris-HCl pH 7.5, 50-500 mM imidazole pH 7.5, 500 mM NaCl, 10% glycerol, 1 mM PMSF). The result of this gradient purification for TgTPS2 is shown in Figure 2A. Each eluted fraction was analyzed by SDS-PAGE (Sodium dodecyl sulfate-polyacrylamide gel electrophoresis): selected eluates were pooled and concentrated using a 10 kDa centrifugal spin column using 25 mM Tris-HCl pH 7.5, 10% glycerol, and 100 mM NaCl as desalting buffer and then aliquoted and flash-frozen for long-term storage at -80°C. Figure 2B illustrates the final result of concentrated protein from such a purification strategy.
(
Purification and enzymatic characterization of TgTPS2
From 1 L of culture, >90% pure TgTPS2 was purified (Fig. 2A). The Bradford assay was initially used for quantification, but exhibited a nonlinear relationship. Therefore, the BCA assay (Pierce kit) was used to provide the required linear relationship for protein quantification according to manufacturer's instructions. A total of 1 mL concentrate was obtained at a concentration of 1 mg/mL, determined by the BCA assay (data not shown).
Biochemical characterization of purified sesquiterpene synthases
Utilization of “cluster tubes” in strips of 8 with accompanying lids in strips of 8 in a 96-well plate format is key to this method. Each strip represents one of the statistically necessary triplicates such that it includes negative and positive controls in addition to the substrate concentrations or other variables to be assayed.
Cluster tubes are normally used for extracting nucleic acids and protein from plants and fungi using a bead-beating principle. They are chemically and physically robust, regularly available at little expense, compatible with the 96-well form factor, and finally, they are more convenient than glass vials.
Controls in the assay were buffer alone, enzyme alone, substrate alone, and finally, samples from expression and purification of a pellet that harbors an empty expression vector.
Varying enzyme and substrate concentrations were tested in a small pilot assay setup to establish the intervals where substrate and enzyme concentration exhibits an adequate measurable activity. For each pilot reaction, 0.1-5 μg enzyme was assayed against 1.625-200 mM FPP, final pH 7.5, spiked with tritium-labeled FPP. This method established the adequate enzyme and substrate concentrations for low reagent usage and short assay time. Each substrate concentration was assayed in mono- or duplicate for one minute, including controls (reaction mix only, boiled enzyme). The pilot assay was performed identically to the final assay as described below.
Following the establishment of optimal enzyme concentration, the final assay could be performed. The enzyme fraction (EF) for one reaction was 1 μg enzyme diluted to 20 μL using 5 mM Tris-HCl pH 7.5, including triplicates with boiled and denatured enzyme. The substrate fraction (SF) for one reaction (80 μL per reaction) was made for each final substrate concentration to be assayed: 200, 100, 50, 25, 12.5, 6.75, 3.375, and 0 μM FPP. Each SF was spiked with 3H-FPP to a final specific activity of 0.082 mCi/mmol. Serial dilution was applied, and everything was mixed and kept on ice, not omitting the additional denatured enzyme and any other controls necessary. EF was buffered with Tris-HCl pH 7.5, and to a final reaction concentration of 45-50 mM MgCl2.
First, 20 μL EF was dispensed to each reaction tube. Then, the reaction was started by adding 80 μL SF. Reactions were rapidly overlaid with 200 μL hexane, and tubes were sealed with lids. At the chosen timepoint(s), reactions were stopped by addition of 100 μL of 0.25 M EDTA, together with 0.5 M KOH or NaOH. The tubes were sealed again and were shaken thoroughly while holding the lids secure for at least one minute in order to ensure maximal extraction of product from the reaction phase. No subsequent extractions were performed. Then, the tubes were placed in a storage cassette, centrifuged for 1 minute at 1500 RCF, and either frozen at -20°C for later analysis or analyzed directly.
For analysis, 50 μL hexane overlay or control was mixed with 200 μL Ecoscint A scintillation fluid in a 96-well scintillation plate and sealed with plastic adhesive film. Plates were counted on a Microbeta 1450 scintillation counter for one minute.
The results were analyzed using Microsoft Excel for calculations of raw data and SigmaPlot 12 or 13 for data analysis and results. The amount of substrate converted in each reaction was calculated, and the amount of enzyme was standardized to per minute per microgram. The reaction rate per microgram enzyme per second was established, and the reaction rate (1/second) was calculated. The triplicate data were processed using SigmaPlot to provide enzymatic parameters, graphs, and error bars.
Results and Discussion
Originally, this protocol was developed for characterizing plant sesquiterpene synthases, specifically from T. garganica. The kinetic parameters obtained are listed in Table 1, and Figure 3 shows that TgTPS2 follows first-order Michaelis-Menten kinetic. Table 1 also presents kinetic data for several plant sesquiterpene synthases obtained over the past three decades. KM for FPP was found to be 0.55 ± 0.3 μM for TgTPS2, Vmax was found to be 0.023 ± 0.001 μM/s, kcat was found to be 0.29 s−1, kcat/KM was found to be 0.53 μM−1 s−1, and [E] was found to be 0.077 μM. These parameters are in a similar range of previously reported data for sesquiterpene synthases as seen in Table 1, where KM and kcat ranges between 0.4 and 23 μM (excluding the two Ginkgo sesquiterpene synthases) and 1.58 × 10−5 to 2.6 s−1, respectively.
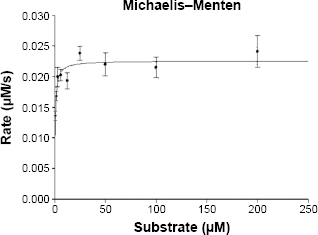
Michaelis-Menten kinetics of TgTPS2. TgTPS2 follows a normal Michaelis-Menten kinetics model. This diagram was obtained by data analysis of scintillation counts using SigmaPlot 13 software.
The higher enzyme parameters achieved when TgTPS2 is expressed and isolated from yeast rather than the common E. coli could be related to secondary modifications of the protein by the two organisms. As shown in Table 1, the parameters of the E. coli and yeast versions of TgTPS2 show that the yeast version requires significantly less substrate to reach KM. Although this observation requires further studies to be elucidated, both results are in the range of what is seen by others. It is common that similar enzymes show different kinetic parameters in different studies as also shown in Table 1, where KM ranges from 0.4 to 8.9 μM for AaADS, KM ranges from 3.4 to 23.3 μM for TEAS, and finally, KM ranges from 0.59 to 1.4 μM for SaSSy. This shows that the kinetic parameters are assay and laboratory dependent, and comparisons require a large dataset background to identify true outliers such as the two Ginkgo sesquiterpene synthases where KM has been found to be 142.9 and 77.42 μM.
The developed method showed significant improvement in throughput compared to our previous studies. 14 We have handled hundreds of samples per day, similar to the MG- and NADPH-dependent assay.15,16 Vardakou et al 15 stated that the MG assay itself (excluding enzyme purification) can be performed in about one hour for up to 30 samples including triplicates, whereas the GC-MS assay takes 8-10 hours including GC-MS runs, 13 and regular radiochemical assay can be completed in 4-5 hours. 14 In the present work, the radiochemical method could be performed in one to two hours. Thus, the presented method measures the sesquiterpene synthase activity as swiftly as the MG- and the NADPH-based methods do. Thus, the current method can be seen as a time-, reagent-, and waste-reducing assay for radiochemical enzyme assays, utilizing a high-throughput format. The main reason for waste reduction is the smaller volume, which reduces reagents and materials required by a factor of 20 compared to our previous work. 14 This altogether reduces the time and reagent use significantly.
This assay utilizes the fact that FPP is water soluble, polar, and will remain in the reaction phase. Any product from the sesquiterpene synthase will be less polar, less water soluble, and will have a tendency to evaporate during the reactions. 20 Therefore, such products must be captured by an organic solvent that is capable of extracting such potential products. We chose hexane due to prior art within the field.14,21–23 Hexane was overlaid on the reaction phase, thus extracting the hydro-phobic products continuously. Final extraction was done by shaking for at least one minute. Pilot extractions indicated that residual products in the reaction and interphase were negligible, and spiking a reaction mix with a known amount of sesquiterpenoid confirmed this. Hexane can emulsify with the reaction phase; this is solved by centrifugation and/or freezing to separate the phases after extraction by vigorous shaking.
Freezing the whole reaction tube directly after extraction does not affect the final scintillation counts, and hexane evaporation during freezing is not a concern. Freezing actually improves the ability to precisely remove the hexane phase for measurements without withdrawing any of the frozen reaction phase, thus improving the reliability and reproducibility of assays. Due to the high volatility of hexane, low freezing temperatures are also able to reduce evaporation during handling of the samples.
FPP may decompose to nerolidol spontaneously or to farnesol by the action of phosphatases. This introduces a risk of measuring false-positive conversions. However, it was observed in prior work that such decomposition was negligible in even longer assays, and the same was observed in this work. Nerolidol is an endogenous product of yeast; 14 thus, empty vectors and fully empty controls should be added to confirm that the yeast protein extracts do not contain carryover of endogenous yeast products such as nerolidol that might interfere with the analysis.
Conclusion
This method demonstrated that sesquiterpene synthases were successfully expressed and purified from yeast at a high level and purity. The enzyme assays demonstrated that TgTPS2, a plant sesquiterpene synthase expressed and purified from yeast is able to convert FPP into its corresponding products and that this conversion is readily detectable by standard analytical chemistry techniques. To the best of our knowledge, this is the first report where plant sesquiterpene synthases are expressed and purified heterologously from yeast in order to perform kinetic assays. The described method reduces waste significantly and avoids glass waste, and final savings on reagents were more than 40%. Throughput was doubled, and reciprocally, time use was reduced to half. Enzymatic parameters were established as follows: KM of TgSTS2 = 0.55 μM; Vmax = 0.023 μM s−1; kcat = 0.29 ± 0.02 s−1; and kcat/KM = 0.53 s−1 μM−1.
Author Contributions
Conceived and designed the experiments: TM and HTS. Analyzed the data: TM. Wrote the first draft of the manuscript: TM. Contributed to the writing of the manuscript: HTS. Agreed with manuscript results and conclusions: TM and HTS. Jointly developed the structure and arguments for the paper: TM and HTS. Made critical revisions and approved the final version: TM and HTS. All the authors reviewed and approved the final manuscript.
Footnotes
Acknowledgment
The authors thank Tom Hamborg Nielsen for valuable discussion prior to the experiments.