
Abstract
Being secondary amines, both salbutamol (SLB) and terbutaline (TRB) react by Maillard reaction (MR) with lactose, which is added as an inactive ingredient in tablets. The Amadori rearrangement products were synthesized, isolated, and characterized by mass spectrometry. In addition, a simple, selective, and precise reversed-phase liquid chromatography (LC) method was developed and validated for the determination of SLB and TRB in tablets, each in the presence of its MR impurity. The chromatographic separation was performed on a Symmetry® Waters C18 column (150 mm × 4.6 mm, 5 μm) using a mobile phase consisting of 0.5% aqueous phosphoric acid to acetonitrile (90:10, v/v) at a flow rate of 0.7 mL minute−1. Quantitation was achieved using UV detection at 230 nm. Linearity, accuracy, and precision were found to be acceptable for the determination of SLB and TRB in the concentration range of 0.2-60 and 0.5-80 μg mL−1, respectively. The proposed method was successfully applied to the determination of SLB and TRB in bulk and in their tablets.
Keywords
Introduction
Salbutamol (SLB), (RS)-4-[2-(tert-butylamino)-1-hydroxyethyl]-2-(hydroxymethyl)phenol (Fig. 1), and terbutaline (TRB), (RS)-5-[2-(tert-butylamino)-1-hydroxyethyl]benzene-1,3-diol (Fig. 2), are two well-known β2-adrenergic receptor agonists used for the relief of bronchospasm in conditions such as asthma and chronic obstructive pulmonary disease. 1
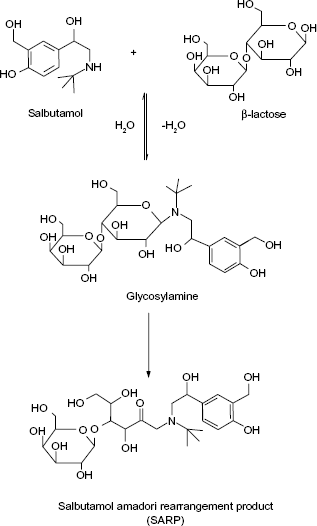
A schematic diagram showing MR between lactose and SLB by refluxing with triethylamine in dimethylacetamide and ethanol.
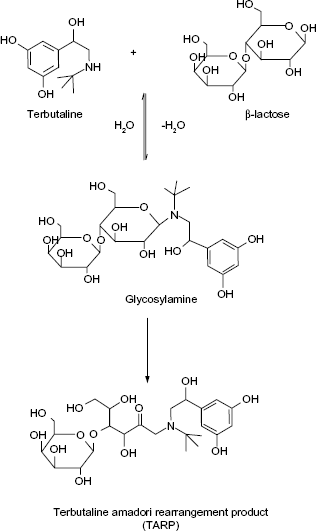
A schematic diagram showing MR between lactose and TRB by refluxing with triethylamine in dimethylacetamide and ethanol.
The reaction between amino compounds and reducing sugars is known as Maillard reaction (MR), and the chemical pathways of the MR are very complicated. The majority of “small molecule” active pharmaceutical ingredient instability reactions occur via hydrolysis, oxidation, and MR. 2 Drugs that are secondary amines, not just primary amines, are reported to undergo MR with lactose under pharmaceutically related conditions.
Hodge presented it as a multistage reaction series in which each stage depends on a number of controlling factors such as temperature, pH, and nature of reactants. 3 High temperature and high pH support a quick completion of the reaction. However, degradation products formed by MR have also been observed under less extreme conditions in pharmaceutical products containing both lactose, an excipient commonly used in solid dosage formulations, 4 and drug substances containing amino groups, such as fluoxetine hydrochloride, 5 aminophylline, amlodipine, acyclovir, 2 and baclofen. 6
Formation of MR impurities was also reported in memantine tablets, 7 and they were simultaneously determined with memantine in tablets using LC with charged aerosol detector. 8
Several analytical methods have been described for the determination of SLB in pharmaceutical and biological samples. These methods include spectrophotometry,9–12 LC,13,14 LC–MS/MS, 15 capillary electrophoresis,16–18 and TLC. 19 TRB has been analyzed by spectrophotometry,20,21 LC,22–25 capillary electrophoresis, 26 and HPTLC. 27
Survey of many generic formulations of SLB and TRB revealed the presence of lactose as the most common excipient, which makes the formulation less stable than those including starch as diluent because of the possibility of MR between each of the two drugs, being secondary amines, and being lactose. Based on this knowledge, the aim of this work was to synthesize MR adducts of both SLB and TRB with lactose (SARP and TARP, respectively) and to elucidate their chemical structures by different spectroscopic techniques. The method of Wirth et al. 5 to prepare fluoxetine adduct was adapted to SLB and TRB. In addition, a sensitive, precise, and accurate LC method was to be developed and validated for the determination of these β2-adrenergic agonists in tablets, each spiked with its MR impurity. A schematic diagram showing the MR between lactose and SLB or TRB is presented in Figures 1 and 2. No reported method has described the separation of the cited drugs from their adduct impurities, which is the aim of the present investigation.
Experimental
Instrumentation
The HPLC system consisted of Shimadzu LC-20AT Liquid Chromatograph (Japan) using a Symmetry® Waters C18 column (150 mm × 4.6 mm, 5 μm). The system was equipped with a UV–visible detector (SPD-20A, Japan) and an autosampler (SIL-20A, Shimadzu, Japan). An Elma S100 ultrasonic processor model KBK 4200 (Germany) was used for the degassing of the mobile phases. Mass spectra were performed on Shimadzu GCMS-QP1000EX mass spectrophotometer (Kyoto, Japan).
Materials and reagents
Pharmaceutical grade SLB, lactose monohydrate, and ventolin tablets, nominally containing SLB equivalent to 2 mg of SLB and 139.8 mg of lactose monohydrate per tablet, were supplied by GlaxoSmithKline Company (Cairo, Egypt). Pharmaceutical grade TRB and Aironyl tablets, nominally containing 2.5 mg of TRB and 97.1 mg of lactose monohydrate per tablet, were supplied by SEDICO Pharmaceutical Company (6 October City, Egypt). Acetonitrile (HiPerSolv) was purchased from Fisher Scientific (Loughborough, Leicestershire, UK). Orthophosphoric acid (85%) was obtained from VWR Chemicals (Pool, England). Bi-distilled water was produced in-house (Aquatron Water Still, A4000D, UK). Membrane filters (0.45 μm) from Teknokroma (Barcelona, Spain) were used.
Preparation of lactose-salbutamol (SARP) and lactose-terbutaline (TARP) amadori rearrangement products
In a 100 mL round-bottom flask, 3 g of lactose monohydrate (8.325 mmol) and 1 g of SLB (4.176 mmol) or 1 g of TRB (3.64 mmol) were dissolved in 50 mL of ethanol. In all, 24 mL of dimethylacetamide and 0.6 mL of triethylamine (4.305 mmol) were added. The mixture was refluxed stirring for 48 hours. The reaction mixture was cooled and filtered, and the residue was washed with 25 mL of ethanol. The filtrate was evaporated under vacuo to a thick solution. A total of 30 mL of ethyl acetate, 30 mL of 20% aqueous sodium chloride, and 10 mL of ether were added. The pH was adjusted to 11 with 50% sodium hydroxide. The layers were separated, and the bottom layer was discarded and the upper layer was set aside. To the middle layer, 30 mL of ethyl acetate and 10 mL of ether were added. The layers were separated, and the combined organic layers were evaporated under vacuo, to which were added 20 mL of 20% aqueous sodium chloride, 40 mL of water, and 60 mL of chloroform. The pH was adjusted to 1.5 with concentrated HCl. The layers were separated, and to the aqueous layer were added 6 g of NaCl, 50 mL of ethyl acetate, and 10 mL of ether. The pH was adjusted to 10.6 with 50% NaOH, and the layers were separated. The ethyl acetate layer was evaporated in vacuum to give a residue of 1.8 g of SARP or 2.3 g of TARP.
Chromatographic conditions
Chromatographic separation was achieved on a Symmetry Waters C18 column (150 mm × 4.6 mm, 5 μm). Isocratic elution using a mobile phase consisting of 0.5% aqueous phosphoric acid and acetonitrile (90:10,
Standard solutions
Standard stock solutions of each drug (1 mg mL−1) were prepared by dissolving 100 mg of the drug in methanol in a 100 mL volumetric flask and then completed to volume with methanol. Standard stock solutions, SARP and TARP impurities (1 mg mL−1), were prepared by dissolving 100 mg of the prepared adduct in methanol in a 100 mL volumetric flask and then completed to volume with methanol. Then required concentrations were prepared by serial dilutions with methanol of these stock solutions. Laboratory-prepared mixtures were prepared by spiking standard solutions of SLB and TRB with their lactose-adducts prepared in Section 2.3.
Sample preparations
A total of 20 tablets of each drug were weighed. An accurately weighed amount of the finely powdered ventolin tablets equivalent to 24 mg of SLB base (28.8 mg SLB) and Aironyl tablets equivalent to 20 mg of TRB were separately made up to 100 mL with methanol. The solutions were sonicated for five minutes to dissolve and filtered followed by serial dilution to the required concentrations for each experiment.
Results and Discussion
Preparation and characterization of sarp and TARP
The MR between SLB and TRB with lactose to produce the SARP or TARP, respectively, is shown in Figures 1 and 2. As reviewed in literature, preparation of Amadori rearrangement product of fluoxetine, which is a secondary amine drug, can be achieved by reaction of lactose with fluoxetine hydrochloride in
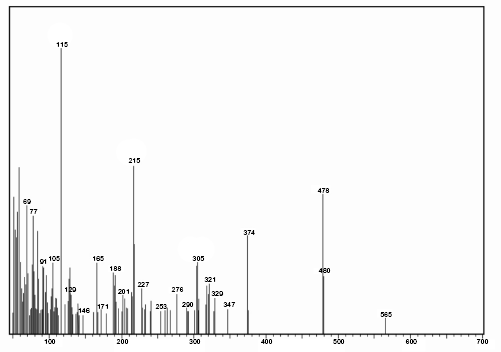
Mass spectrum of SARP.
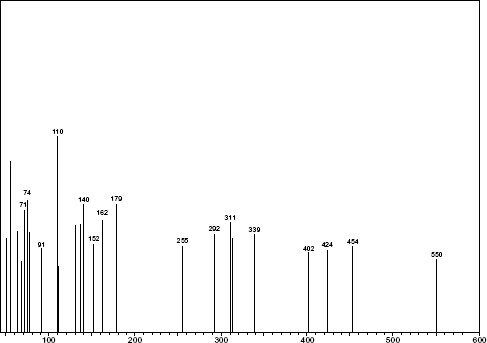
Mass spectrum of TARP.
LC Method development
During the optimization cycle, various reversed-phase columns, isocratic mobile phase systems, and pH values of the buffer were attempted. To detect each of SLB and TRB and their MR impurity with sufficient peak intensity, a wavelength of 230 nm was selected. It was found that at least 90% of aqueous phase was needed to elute all peaks within 10 minutes because of relatively high polarity of the drugs and their lactose-adducts. Isocratic elution based on a mobile phase consisting of 0.5% aqueous phosphoric acid and acetonitrile (90:10,
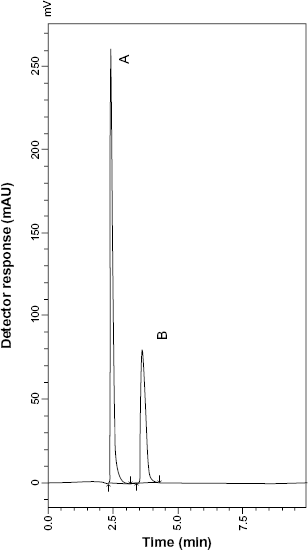
HPLC chromatogram of SLB sulfate (10 μg mL−1) (A) and SARP (3 μg mL−1) (B).
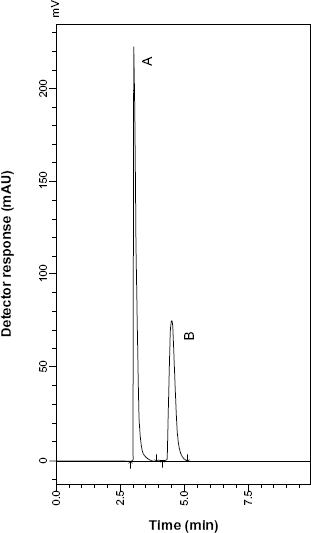
HPLC chromatogram of TRB sulfate (10 μg mL−1) (A) and TARP (3 μg mL−1) (B).
System suitability tests
System suitability tests are an integral part of liquid chromatographic methods in the course of optimizing the conditions of the proposed method. 28 %RSD of peak area for six injections of a solution of 30 or 40 μg mL−1 (100% concentration) for SLB or TRB, respectively, was used to test repeatability, and%RSD of retention time to test retention time reproducibility. The results of these tests for the proposed method are listed in Table 1. Drugs under investigation were well resolved from their MR impurities as shown in Figures 5 and 6. Resolution factors were found to be 2.404 between SLB and SARP and 2.403 between TRB and TARP.
System suitability tests for LC method for the determination of SLB and TRB in the presence of their MR impurities.

Method validation
Linearity. In this study, eight concentrations were chosen for each drug. Each concentration was repeated three times. Good linearity of the calibration curves was verified by the high correlation coefficient (Table 2).
Results obtained by LC method for the determination of SLB and TRB in the presence of their MR impurities.

Accuracy
Accuracy of the results was calculated by percentage recovery of five different concentrations of SLB and TRB standard solutions spiked with different quantities of SARP and TARP impurities in standard solutions (3-5 and 3-7 μg mL−1), respectively (10-30%) (w/w). Triplicate sample preparations were made at each concentration level. The results obtained including the mean of the recoveries and standard deviation are displayed in Table 2 indicating good accuracy of the proposed method.
Precision
For SLB and TRB, repeatability of the method was assessed by analyzing a standard solution containing 30 and 40 μg mL−1, respectively (n = 6). In addition, intra-day and inter-day precision (using three different concentrations in triplicates for three consecutive days) was also carried out. The three concentrations applied are (24-30-36 μg mL−1) for SLB and (32-40-48 μg mL−1) for TRB, representing 80-100-120%, respectively. The values of%RSD of repeatability and intra-day and inter-day precision were all found to be less than 2%. The results are displayed in Tables 1 and 2.
Specificity
Good resolution and absence of interference from any of the MR impurities are shown in Figures 5 and 6. Also, the chromatograms of the pharmaceutical formulation samples were checked for the appearance of any extra peaks. No chromatographic interference from any of the excipients was found at the retention times of each of the examined drugs after extraction of the active ingredient (Figs. 7 and 8). In addition, the chromatograms of SLB and TRB in the sample solution were found to be identical to the chromatogram obtained by the standard solutions.
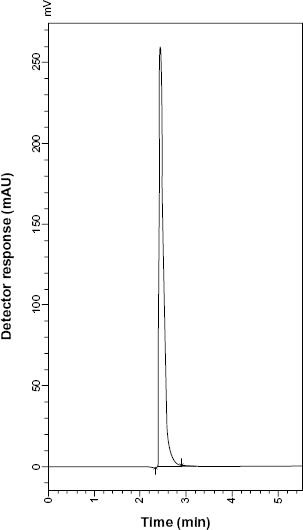
HPLC chromatogram of ventolin tablets containing 10 μg mL−1 SLB sulfate.
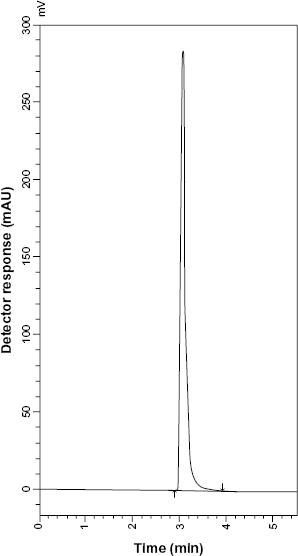
HPLC chromatogram of Aironyl tablets containing 10 μg mL−1 TRB sulfate.
Limit of detection (LOD) and limit of quantitation (LOQ)
Limit of detection that represents the concentration of analyte at
Robustness
Robustness was performed by deliberately changing the chromatographic conditions. The flow rate of the mobile phase was changed from 0.7 to 0.6 and 0.8 mL minute−1. The organic strength was varied by ±2%. It was found that these variations did not affect the chromatographic resolution significantly when the proposed LC method was applied for the determination of SLB and TRB, each in the presence of its Maillard impurity.
Conclusion
The MR products of SLB and TRB with lactose were synthesized, and their chemical structures were elucidated. The proposed LC method has the advantages of simplicity, precision, accuracy, and convenience for the separation and quantitation of SLB and TRB. In addition, it is suitable for the simultaneous determination of their binary mixtures with their MR impurities. Hence, the proposed LC method can be used for the quality control of the cited drugs in ordinary laboratories.
Author Contributions
Conceived and designed the experiments: MF, AE, EE. Analyzed the data: MF, AE, EE. Wrote the first draft of the manuscript: MF, AE, EE. Contributed to the writing of the manuscript: MF, AE, EE. Agree with manuscript results and conclusions: MF, AE, EE. Jointly developed the structure and arguments for the paper: MF, AE, EE. Made critical revisions and approved final version: MF, AE, EE. All authors reviewed and approved of the final manuscript.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.