
Abstract

Editor:
We appreciated the quality of the project by Anjum et al.,[1] concerning circulating endothelial progenitor cells (EPCs) in patients with sickle cell disease. An increasing body of data indicates that intravascular hemolysis contributes to a state of vascular dysfunction in sickle cell disease, especially involving diminished nitric oxide (NO) bioavailability. This vascular dysfunction is detected on venous occlusion plethysmography blood flow physiology studies as a relative blunting of NO-induced vasodilation[2–4] and this state of NO resistance has also been observed in sickle cell mouse models.[5,6] This vascular dysfunction may play a role in the development of pulmonary hypertension in sickle cell disease.[7]
In our study of 24 adults with sickle cell anemia (SCA), EPC levels trended lower than in 10 healthy African-American controls (median 2.50 vs. 8.88 colonies/well, P = 0.059, Fig. 1A). This difference might be due to SCA, but also possibly to the study entry criteria for the SCA subjects that selected mainly for patients with high soluble vascular cell adhesion molecule-1, a marker associated with vascular dysfunction.[8] Low circulating counts of EPCs have correlated in the general population with cardiac disease risk factors,[9] supporting parallels between markers of vascular disease in the general population. Consistent with such a parallel, we found that lower than median EPC number in our SCA subjects is associated with elevated Doppler-estimated pulmonary arterial pressures by tricuspid regurgitant velocity (mean 2.57 vs. 2.38 m/s, P = 0.039, Fig. 1B). We also find that low EPC number is associated with endothelial dysfunction, detected as a blunted vasodilatory response to acetylcholine infusion into the brachial artery (P = 0.017, Fig. 1C), but not with infusion of sodium nitroprusside as a control for endothelial-independent vascular function (Fig. 1D).
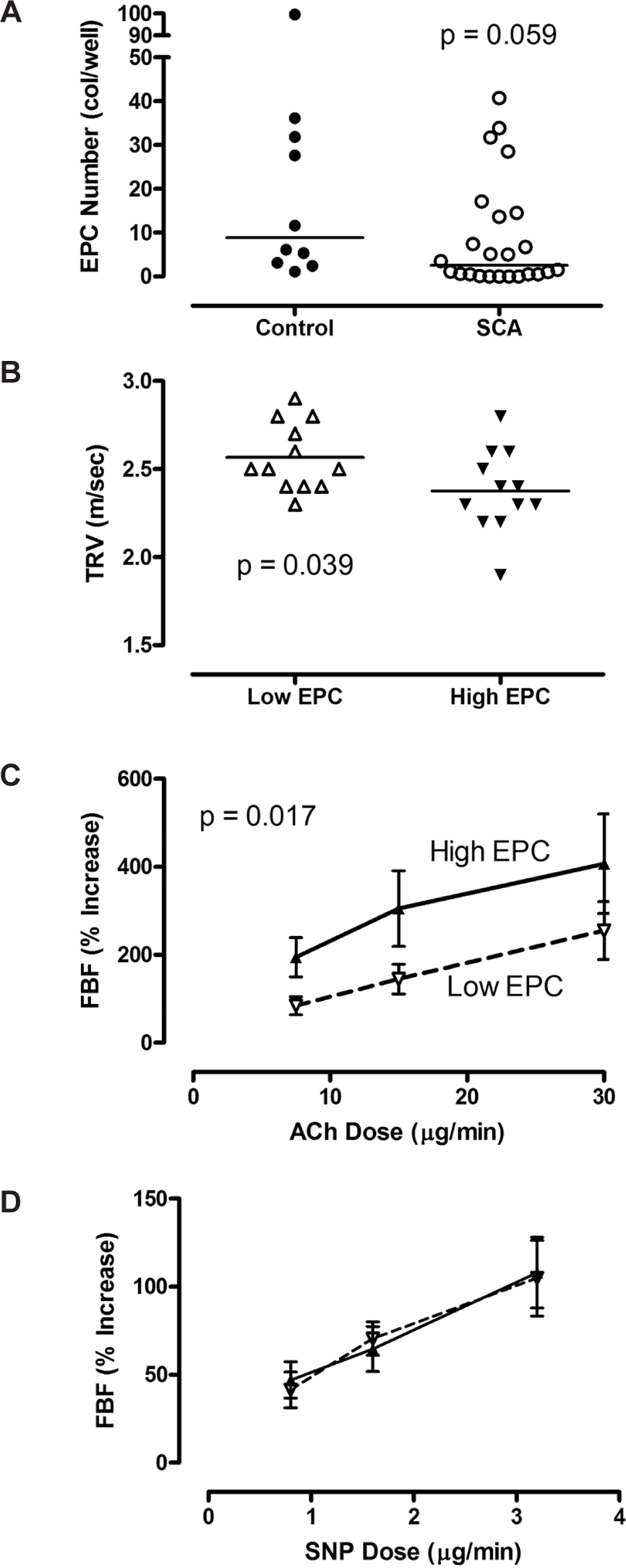
Endothelial progenitor cells in sickle cell anemia (SCA) and control subjects.
We note with caution the risk that statistical significance by Anjum et al.,[1] could be an artifact of the multiple comparisons they computed. Similarly, we note caveats with our own results. To measure circulating EPC, we used a fibronectin cell culture assay that was previously accepted. Our own gene expression profiling publication suggests contamination of the EPCs detected in that assay with activated T lymphocytes, clouding accurate quantification by the cell culture EPC assay.[10] However, the concurrence of EPC results and elevated pulmonary artery pressure from our two research groups using different assay approaches suggests that the findings of both groups are not simply artifacts, and may reflect mutually consistent findings. Additional research in this area is needed.