
Abstract
To catch an imagination of the future of pulmonary hypertension was exactly the spirit of the 55th ASPEN lung Conference. Basic scientists, pre-clinicians, clinicians and pharma joined together to achieve one goal—to combine creativity and inventiveness in a battle against a deadly disease. Summarizing this conference on “Mechanics and Mechanisms of Pulmonary Hypertension” is challenging in several aspects: To extract key novel findings from 12 state-of-the-art lectures, 25 oral presentations, 56 posters along with the integration of own data on discussed topics, to include hundreds of important questions, answers and discussion raised during the conference, to provide the line of thinking for the next 5–10 years of pulmonary hypertension (PH) research development and to focus equally well on both basic and translational research. Kurt Stenmark and Todd Bull, who chaired the conference, intensified this challenge several-fold by selecting a plethora of topics ranging from development of cardiopulmonary systems to pathogenesis of right ventricular failure, mechanics of right ventricle-pulmonary artery coupling to genomics and from understanding metabolic aspects to developing therapies for PH. With that, need not say, but a special admiration and thanks to the conference chairs for assembling such outstanding state-of-the-art speakers, for clustering the presentations logically and for leading lively and engaging discussions. Although it may look fragmentary, we would like to divide the conference summary into four major conceptual realms: The pulmonary vasculature in PH; right heart in PH; individualized approach- personalized medicine; and beyond PH-vascular abnormalities in COPD.
- Joseph Joubert
THE PULMONARY VASCULATURE IN PH
The development of therapeutic concepts in PH was intimately linked with the unraveling of pathogenetic aspects contributing to this devastating disease. Though the molecular pathogenesis driving the abnormal inward remodeling processes in the different PH entities is still largely unknown, several genetic, molecular and cellular abnormalities have been identified as important players in the pathogenetic sequelae, thereby also offering as targets for therapeutic intervention.
Imbalance of vasomotion
Central role of endothelial dysfunction in the initiation and progression of PH resulted from altered production of endothelial vasoactive mediators. Of those mediators, nitric oxide (NO), prostacyclin (PGI2), serotonin (5-HT) and endothelin (ET) are among the best studied and resulted in three currently approved therapeutic strategies for the treatment of pulmonary arterial hypertension (PAH); PGI2 and analogues (iloprost, treprostinil),[1,2] PDE5 inhibitors (sildenafil, tadalafil),[3,4] and ET receptor antagonists (bosentan, ambrisentan)[5,6] have emerged in part from restoring the balance between these mediators. Current developments in these established and validated pathways are summarized in Figure 1. Preserving the advantageous effects of prostacyclin, while avoiding several of its limitations upon systemic delivery, the following chain of translational development is currently under investigation: (1) Aerosol technology (metered dose concept for iloprost and treprostinil); (2) Controlled release formulations for aerosol delivery (prostanoids packaged in liposomes and nanoparticles); and (3) clinical trials with oral PGI2 receptor agonist, selexipag (GRIPHON study, recruitment ongoing). With regard to endothelin signaling, the clinical trial with the oral nonselective endothelin A/B antagonist macicentan (SERAPHIN study) was recently finished and reported to be positive as based on press release. Extending therapeutic options involving NO downstream signaling pathways employs the pharmacological activation of soluble guanylate cyclase (sGC) by compounds like riociguat. Recent studies have shown that riociguat elicits strong pulmonary vasodilatation and reverses remodeling in experimental PH.[7] Phase II trials with riociguat in PAH and chronic thromboembolic PH (CTEPH) have demonstrated clinical benefits,[8] and two Phase III trials in PAH and CTEPH (PATENT and CHEST) have recently been finalized, with results to be expected in the near future. In addition, experimental evidence for therapeutic efficacy of PDE isoforms other than PDE5, PDE3 and PDE4,[9] as well as nitrite (Diana Tabima) was claimed: The latter agent elicits several “metabolic” effects and causes NO liberation, offering for both systemic and inhalative administration.
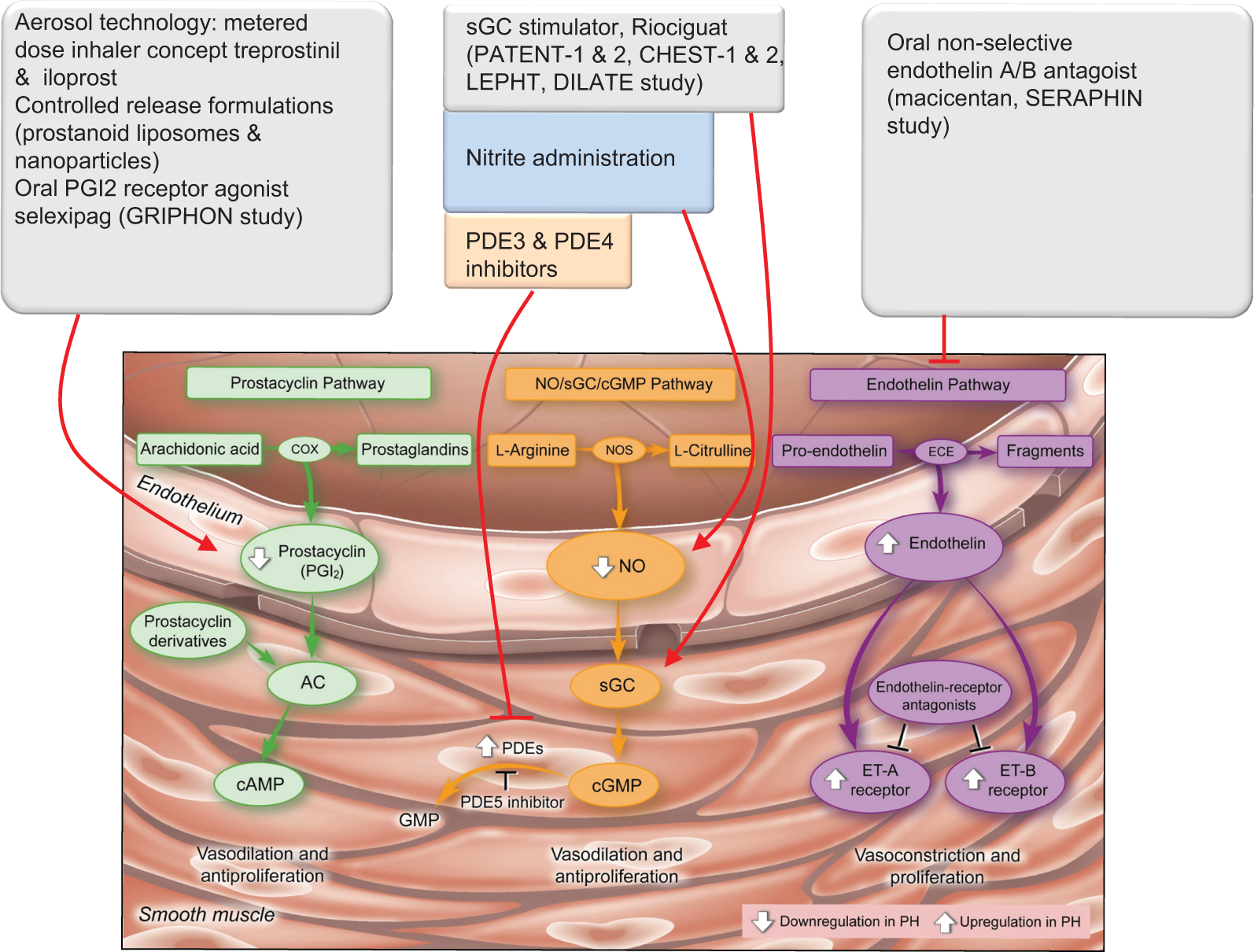
Current clinical developments in established and validated pathways in PH. COX: Cyclooxygenase; AC: Adenylate cyclase; cAMP: Cyclic adenosine monophosphate; NOS: Nitric oxide synthase; NO: Nitric oxide; sGC: Soluble guanylyl cyclase; cGMP: Cyclic guanosine monophosphate; PDE: Phosphodiesterase; ECE: Endothelin converting enzyme; ET-A: Endothelin receptor A; ET-B: Endothelin Receptor B
Hyperproliferative disease—paradigm shift
Hitherto, current PH therapy provides symptomatic relief and improves prognosis, but falls short as to re-establishment of structural and functional lung vascular integrity, as a basis for handicapped free long-term survival. Moreover, by orders of magnitude more frequent, the unmet clinical need is even more pronounced in pulmonary vascular disorders outside the PAH group, e.g., those with underlying heart or lung disease and CTEPH, for all of which no single medical treatment is currently approved. Hence deciphering the molecular mechanisms, which drive the maladaptive inward remodeling processes in PH, is indispensable. The restoration of physiological vascular structure and function (reverse remodeling) represents the therapeutic goal. This requires better understanding of the switch from “quiescent” to “proliferative” cell phenotypes, disruption of vicious pathogenic circuits, which drive angioproliferative abnormalities and activation of repair and regenerative mechanisms.[10,11] The following sections represent a broad assessment of a complex set of mechanisms that we felt should represent important areas for focused research and for the development of reverse-remodeling strategies. Different approaches undertaken to achieve this goal are summarized in Figure 2.
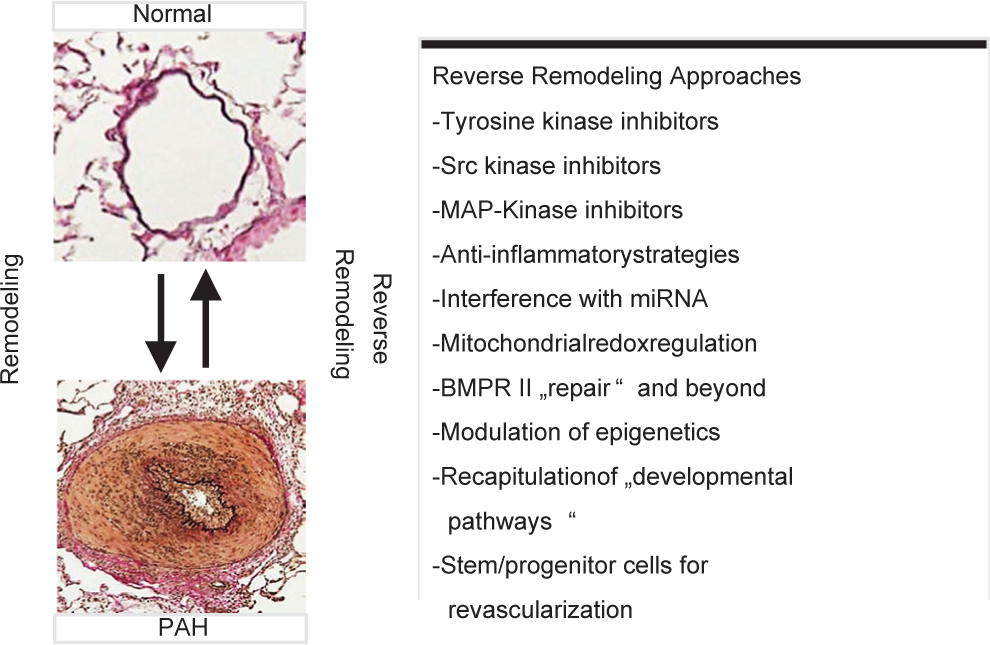
Different strategies addressing remodeling and reverse-remodeling in PH. MAP-kinase: Mitogen activated protein kinase; miRNA: microRNA; BMPR II: Bone morphogenetic protein receptor, type II
Tyrosine kinase and Src kinase inhibitors
Ralph Schermuly and coworkers first demonstrated that platelet-derived growth factor (PDGF alpha) plays a pivotal role in the “pseudomalignant” inward remodeling of PH lung vessels via receptor tyrosine kinase (RTK)-dependent signaling.[12] Inhibitors of RTK enzymes, in particular imatinib, reversed the pathological remodeling in experimental PH and imatinib exhibited promise in a Phase II trial.[13] A multicenter Phase III trial of imatinib in PAH (IMPRES) was recently finished (Ardeschir Ghofrani). Imatinib significantly improved 6-Minute Walk Distance, cardiac output, pulmonary vascular resistance and NT-pro-BN P values. However, adverse effects were higher in the imatinib group as expected for this class of drug and as a surprise increased number of subdural hematoma was noted. Approval of imatinib for treatment of PAH is currently under investigation by the FDA and the EMEA.
In addition, activation of epidermal growth factor (EGF), fibroblast growth factor (FGF), insulin like growth factor (IGF) and insulin receptors was shown to contribute to pulmonary arterial smooth muscle cell (PASMCs) proliferative responses.[14,15] These findings suggest that inhibitors that can target a common downstream signaling molecule of RTKs can be of potential therapeutic value. Such reasoning definitely left us at a cross-road of multiple ways: (1) To take a broad spectrum multikinase inhibition approach (e.g., sorafenib and sunitinib); (2) to screen for inhibitors that can target a common downstream signaling molecule of RTKs (e.g., Src kinase); or (3) to strive for a narrow focus of RTKs to increase the safety, specificity and efficacy of this approach. Definitely, treatment with multikinase inhibitor sorafenib showed reverse-remodeling efficacy in experimental models of PH.[16] Similarly, Soni Savai Pullamsetti showed that dasatinib, a dual inhibitor of PDGFR receptor and Src kinases, potently inhibits mitogenic and motogenic responses to growth factors in PASMCs and pulmonary vascular remodeling in vivo.[17] To much surprise, dasatinib in clinical use in oncology was noted to induce severe precapillary PH fulfilling the criteria of PAH in a small percentage of patients treated with this agent, possibly due to vasoconstrictive/vasoproliferative effects mediated by Src kinases.[18] Thus, the obvious question is how to harness the powerful antiproliferative potency of tyrosine kinase inhibitors to achieve reverse remodeling and improved survival in PH while avoiding severe adverse effects. Shall we focus on local delivery by employing inhalative application, as currently undertaken in lab studies; shall we strive for enhanced potency/selectivity by new generation imatinib analogues (e.g., nilotinib, being currently tested in Phase II trials in PAH); or shall we readdress the question of how to design a tyrosine kinase inhibitor as specific for the molecular signaling underlying P(A) H as possible? Not to be biased for PDGF signaling, but these suggestions apply to other TK signaling pathways as well.
MAP kinase inhibitors
Mita Das provided compelling evidence that gene deletion of JNK1/2 blunts vascular remodeling in hypoxia induced PH, suggesting reverse-remodeling efficacy of JNK kinase inhibitors. Though no further data on MAP kinase inhibitors in PH was presented in this conference, currently undergoing studies focusing on interference with p38 and other MAP kinases were addressed in the discussion.
Inflammatory component in PH- anti-inflammatory strategies
Abundant research over the past decade has implicated inflammation in several categories of PH. Initially, these data focused on characterizing inflammatory cell infiltrates as well as the proinflammatory cytokines/chemokines that are detected in patients with PAH.[19] Marc Humbert summarized these findings and the contribution of inflammatory sequelae and (subsequent) autoimmunity to pulmonary vascular remodeling in different subgroups of PAH patients (idiopathic PAH [IPAH] and connective tissue disease-associated PAH). Up to one-third of PAH patients have circulating autoantibodies against various vascular self-antigens. He provided a structural basis for this local autoimmune response. Recent work from his group demonstrated that perivascular lymphocytic infiltrates could form bona fide highly organized ectopic (tertiary) lymphoid follicles and provided important insights in the cellular and molecular actors involved in initiation, maintenance and function of these perivascular lymphoid follicles.[20] Carrying this observation further in experimental studies, Michael Yeager demonstrated the appearance of ectopic lymphoid tissue in association with PH development and showed that this lymphoid tissue displays unique cellular consistency and gene expression permissive for production of autoantibodies. Interestingly, in vivo pharmacological and immune-based blockade of lymphoid tissue effectors causes drastic influence on pulmonary vascular remodeling: Chemokine receptor type 7-Ig worsens monocrotaline induced PH and is associated with phenotypically altered ectopic lymphoid tissue, whereas lymphotoxin beta receptor-Ig prevents and reverses PH and is associated with decreased ectopic lymphoid tissue. Thus, new anti-inflammatory treatment concepts of PH may show up at the horizon.
Subgroups of PAH in which infection and immunity play a major role include human immunodeficiency virus (HIV) infection and schistosomiasis-associated PAH.[21] HIV infection (> 40 million people affected worldwide, with < 50% in Sub-Saharan Africa) increases the risk for PH development < 1,000-fold, a statistic that remains unchanged even under combination antiretroviral therapy. Sonia Flores presented her impressive studies on chimeric SIV-HIV-nef infected Rhesus macaques to study PH and vascular remodeling in the context of HIV infection. Her data suggest that specific HIV nef alleles are essential for the vascular remodeling and predict disease progression in individuals at risk; furthermore, these alleles may trigger a cascade of events leading to the selection of a rapidly growing, apoptosis-resistant endothelial cell population.
Schistosomiasis is the third leading endemic parasitic disease in the world, with < 300 million infected individuals and an estimated 5–15 million individuals have schistosomiasis-associated PAH.[21,22] Data presented by Brian Graham showed evidence for pathogenic mechanisms linking infection and immune responses to vascular remodeling. Mice experimentally infected with schistosomiasis mansoni have multiple features consistent with PAH, including pulmonary vascular remodeling sequelae. Importantly, these sequelae appear to be dependent on enhanced IL4/IL13 and subsequent TGF-β signaling, suggesting new opportunities for therapeutic intervention. Again focusing on anti-inflammatory pathways, Mark Nicolls showed that targeting eicosanoid-mediated inflammation reverses fulminant PAH in animal models, thus offering a further perspective of intervention. Thus, all these contributions strongly suggest that inflammation plays a causative role in a subset of patients with PAH and that novel anti-inflammatory strategies targeting central mechanistic processes within the inflammatory signaling cascades are worth being developed but may differ between the various inflammation driven P(A) H subtypes.
Modulation of epigenome and miRNome
MicroRNAs (miRs) control various cellular processes in tissue homeostasis and disease by regulating gene expression on the post-transcriptional level. Recently, some of the miRNAs such as mir17-92 cluster and miR21 were shown to be altered in PH. Soni Savai Pullamsetti evaluated the therapeutic efficacy and antiremodeling potential of miR inhibitors (antagomirs) in the pathogenesis of PH. She demonstrated that A-17 improves heart and lung function in two different experimental models of PH by interfering with lung vascular and right ventricular remodeling.[23] Thus, inhibition of miR17, linked with both bone morphogenetic protein receptor type II (BMPRII) signaling and p21 meditated cell cycle control, may represent a novel therapeutic concept. As microRNA inhibitors are currently tested for different indications in various clinical trials, the identification of microRNAs as novel therapeutic targets may offer new approaches for the treatment of PH.
Epigenetic programming, dynamically regulated by histone acetylation, is a key mechanism regulating cell proliferation and survival. Little is known about the contribution of histone deacetylase (HDAC) activity to the development of PAH. Kurt Stenmark's group demonstrated that therapies specifically targeting abnormalities of HDAC activity in adventitial fibroblasts appear to hold promise.[24] In accordance, Lan Zhao showed that pan-HDAC inhibitors, VPA and SAHA prevented and reversed hypoxia-induced PH, thus offering a novel therapeutic strategy. Although modulation of epigenome and miRnome seems promising, the fine details of histone modifications and the causal role of histone modifiers need further in-depth analysis.
Mitochondrial redox regulation
Stephen Archer provided insights on the metabolic remodeling of the right ventricle and pulmonary circulation in PH. He provided evidence for an activation of hypoxia inducible factor 1 alpha (HIF-1α) under aerobic conditions in PH, being associated with a shift from aerobic mitochondrial energy metabolism to aerobic glycolysis. Interestingly, promoting aerobic mitochondrial energy metabolism with the pyruvate dehydrogenase kinase inhibitor dichloroacetate (DCA) has substantial protective effects against PAH and restores RV function in experimental models,[25] and Phase II clinical trials with DCA are currently underway. Recently, his group also found that metabolic shift in PAH is associated with mitochondrial fragmentation, again linked to HIF-1α,[26] and that targeting mitochondrial fission and fusion offers a novel antiproliferative strategy in PAH.
Vascular pruning-regenerative treatment strategies
In addition to the pathological inward remodeling, loss of precapillary vessels (pruning) and impaired vascular regeneration contributes to the severe reduction of pulmonary vascular cross-sectional diameter in PAH. This pruning is assumed to result from increased endothelial cell apoptosis, but the cellular and molecular mechanisms responsible for vessel loss are largely unresolved. Our group recently showed that asymmetrical dimethyl arginine (ADMA), an endogenous NO synthase inhibitor and its metabolizing enzyme dimethyl arginine dimethylaminohydrolase (DDAH), play important roles in endothelial dysfunction. Importantly, combined inhibition of PDE3 and 4 regresses development of pulmonary hypertension and promotes endothelial regeneration and lung vessel density by modulating the cAMP-ADMA-DDAH axis.[9] Further analyzing the cellular and molecular players of vascular pruning, Ke Yuan reported that abnormal pericyte function as well as impaired pericyte and endothelial cell interaction due to loss of Wnt5a signaling leads to small vessel loss and reduced angiogenesis in IPAH. This finding supports the notion that targeting Wnt/planar cell polarity (PCP) pathway may be employed to regenerate pulmonary vessels.
Along the same line, Marlene Rabinovitch discussed specific issues regarding induced pluripotent stem cells (iPSCs). iPSCs may offer a means for induction of angiogenesis in PH within the remnants of obliterated vessels in the diseased lungs. She gave us insights on how pulmonary hypertension breakthrough initiative (PHBI) network is planning to generate iPS cells, either from skin fibroblasts or from pulmonary arterial endothelial cells (PAECs) from IPAH patients, to study the role of these cells in pathogenesis as well as their use as cell-based therapies. These cells will be compared using deep sequencing modalities (exome sequencing, methyl seq, RNA seq) and the differences will be related to angiogenic function. Interestingly, RNA seq gene expression changes in IPAH patients are reflective of abnormal BMPR2 function and additional perturbations that further compromise EC function. Importantly, novel drugs such as elafin and PPAR gamma agonists may target such perturbations and improve EC function. Along the same line, Darwin Prockop provided data on how mesenchymal stem cells and/or the therapeutic proteins they produce can be used for development of stage-directed therapies for PH.
BMPR2 repair and beyond
BMPR2 mutations may lead to reduced BMPR2 expression, which is causally linked to the majority of cases in familial PAH (FPAH) and a substantial percentage of IPAH.[27,28] In addition, reduced expression of BMPR2, even without a loss of function mutation, seems to be a feature of many forms of PAH.[29] Pravin Sehgal showed that IPAH-derived mutant BMPR2 species inhibit endoplasmic reticulum to Golgi to plasma membrane trafficking, linked with downregulation of signal transducer and activator of transcription (STAT5a/b) and endothelial NOS (eNOS), stressing the importance of subcellular mechanisms which may bridge BMPR2 mutations with the PAH phenotype. In an attempt to promote BMPR2 related pathways, Edda Spiekerkoetter performed a high throughput screen to identify appropriate drugs and found FK506 (Tacrolimus) as the best hit activating BMPR2 signaling and reversing related dysfunctional pathways in PAECs from PAH patients. Importantly, in a rat model of severe PH, FK-506 reversed PH and lead to regression of occlusive neointima lesions, suggesting the therapeutic efficacy of FK-506.
Recapitulation of developmental pathways
Aberrant activation of a developmental pathway is a common and well-investigated feature of cancer, but this mechanism is less explored in PH. Edward Morrisey's talk elegantly addressed this topic. He revealed several stunning facts about the co-development of the cardiopulmonary organ system such as that the pulmonary vasculature arises from the developing heart even in the absence of lung development. Moreover, his group noted an intimate crosstalk between Wnt and Sonic hedgehog (Shh), mandatory for development of the pulmonary vasculature. The next obvious question is whether these molecular mechanisms are recapitulated in PH. Work from Edward Morrisey, our group and Marlene Rabinovitch's group has indeed shown that Wnt signaling is aberrantly activated in IPAH vasculature and may contribute to disease pathogenesis.[30–32] Modulation of the Wnt pathway may, therefore, present as a suitable and promising novel therapeutic strategy to reverse aberrant remodeling and to foster morphologically adequate regeneration of the lung vasculature.
RIGHT HEART IN PH
The holy grail in PH is right ventricular (RV) function that determines the survival of the patients.[33] Right ventricular hypertrophy (RVH) in response to increased afterload with subsequent progression to right heart failure (RVF) occurs under various conditions of PH and is also secondary to left heart failure. The ability of the RV to cope with chronic pressure overload strongly depends on its developmental stage and yet unknown individual parameters. The first steps toward a strategy to counteract adverse remodeling events in the RV include progress to assess right ventricular function independent of changes in afterload, improved animal models mimicking right ventricular abnormalities in PH and the identification of promising target molecules and/or regulatory circuits for therapeutic interference (Fig. 3).
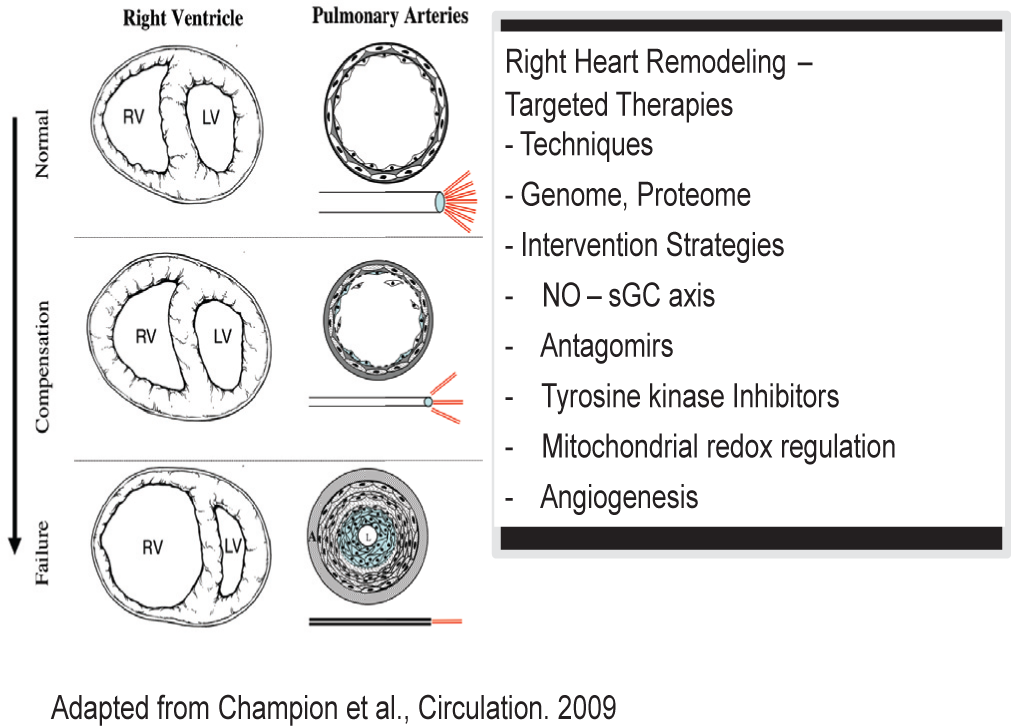
(Left) Schematic showing the theoretical progression of pulmonary vascular disease and the subsequent effect on RV function from normal physiological conditions (top) to severe pulmonary vascular remodeling and subsequent RV failure (bottom). (Right) Schematic representation approaches undertaken to address right heart remodeling and failure. NO: Nitric oxide; sGC: soluble guanylate cyclase.
Along this line, Hunter Champion took us to the intersection/interaction of the right ventricle and the pulmonary circulation-an important aspect that is largely understudied. He highlighted the future of integrative assessment of ventricular-pulmonary vascular coupling via hemodynamic measures. He presented compelling data on how the novel hemodynamic parameters such as assessment of the pulmonary arterial pressure waveform and RV pressure volume loop relations can be useful to the clinical management of PAH patients.[34] He reiterated that the elastic properties of the pulmonary circulation and impedance on RV performance should be considered, rather than the pure resistive properties. This talk emphasized that RV contractility studies are not only relevant to the clinical management of PAH patients but also critical for the interpretation of data from clinical trials as well.
Employing various experimental approaches, Norbert Voelkel provided deep insights into the cardiac functional as well as cellular changes in the RV which leads to RVF.[35] He showed that peroxisome proliferator-activated receptor (PPAR) signaling is differentially regulated in the RV of normal versus hypertrophied RV versus failing RVs. He provided evidence for the role of PPAR and PPAR coactivator-1-alpha (PGC-1α) in mitochondrial dysfunction of the failing RV. The activating effects of PGC-1α on mitochondrial gene expression are exerted through its coactivation of nuclear respiratory factor (NRF1 and NRF2) as well as the estrogen-related receptor α (ERRα). The induction of NRF by PGC-1α results in increased expression of mitochondrial transcription factor A (Tfam), a nuclear-encoded transcription factor essential for replication, maintenance and transcription of mitochondrial DNA. This signaling pathway may thus offer to improve mitochondrial function, energy metabolism and thus performance of the afterload challenged right ventricle in PH.
Addressing the important question, whether absolute or relative loss of capillary density contributes to RV failure in PH, Rubin Tuder presented data from a stringent stereological approach. Employing this technology in rats treated with SU5416 + hypoxia, he observed no reduction of RV capillaries. He thus stressed that at least in this model loss in vessel density does not appear to be a major contributor to the pathogenesis of RVF and that, in general, sound stereological technology is mandatory for assessment of the cardiac vasculature.
Several further talks summarized recently published as well as novel studies in the regulatory gene networks in the hypertrophied and failing RV. In genome-wide gene expression studies undertaken in murine models of pulmonary artery banding versus aortic banding, we identified a substantial number of genes that are differentially regulated in the RV compared to the LV, including those with hitherto unknown cardiac function such as the cartilage intermediate layer protein (CILP), which increases < 20-fold in the mechanically stressed right ventricle.[36] Thomas Di Salvo presented LV-RV expression changes upon ventricular failing in humans, thus providing a large list of genes offering for discrimination between the two ventricles and future therapeutic interference. Rachel Damico elegantly demonstrated that collagen 18a1 (Col18a1) mRNA, a precursor of endostatin, is increased in murine models of RVF. Most importantly, polymorphisms in col18a1 are associated with altered cardiac output in SSc-PAH patients, thus underlining the translational relevance of this finding. Furthermore, preliminary data from our group point to a re-expression of neonatal genes, e.g., Wnt signaling molecules, in the pressure-overloaded right ventricle.
That being said, we should lay pillars on basic research work for right heart focused therapies in PH. The treatment of PH currently includes endothelin receptor antagonists, PDE inhibitors and prostanoids. However, it is not clear whether these drugs affect right heart function and structure independent of an afterload decrease, even though this knowledge might be crucial with regard to long-term patient outcome. In addition, current preclinical findings favor adrenergic receptor blockade, various tyrosine kinase inhibitors as well as sGC stimulators, as pharmacological approaches for direct therapeutic impact on the right ventricle; however, such findings need further verification in different experimental models as well as clinical trials in PH patients particularly assessing right ventricular function.
INDIVIDUALIZED APPROACH-PERSONALIZED MEDICINE
In PH, current assessment of a patient's prognosis and therapeutic decision is largely based on averaged values from studies in large patient cohorts. This fails to take individual heterogeneity into consideration. However, though new therapies for PH emerged over the past years and the individual patient's response to the different strategies varies considerably, such responsiveness is currently not predictable. Our failure to make any substantial progress in this field over the past years is even more frustrating, as new treatment concepts may have serious side effects, demanding critical outweighing of risks and benefits. Current strategies to overcome this shortcoming include genomics, transcriptomics, proteomics, imaging techniques and classical biomarker analysis as summarized in Figure 4.
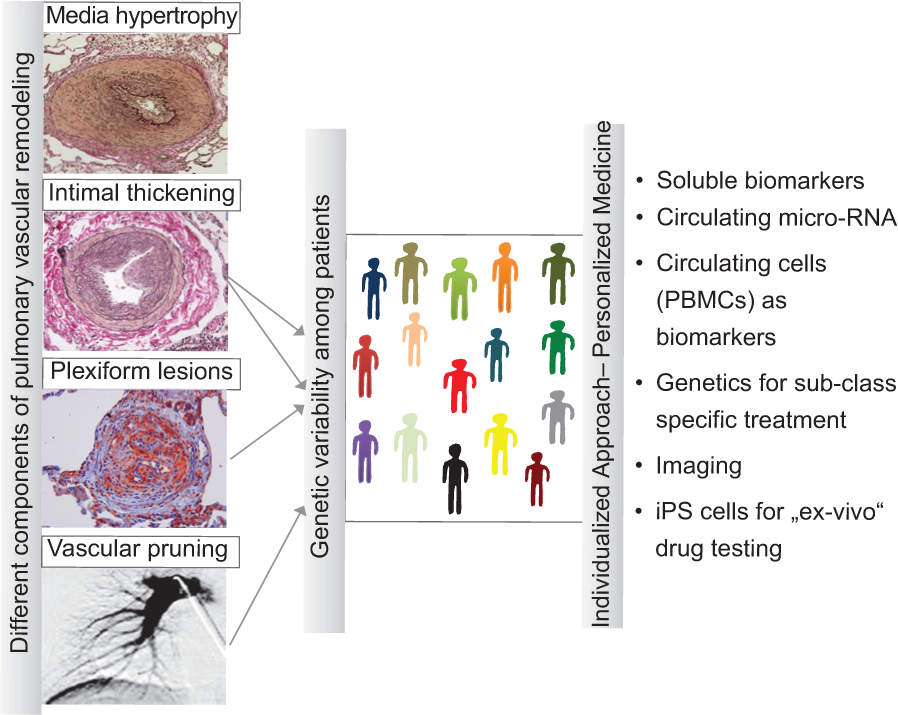
Current strategies to address variability (genetic, molecular, cellular and response to therapy) among PH patients - Personalized medicine.
Against this background, Mark Geraci walked us through the spectrum of genomic approaches (exome sequencing, epigenomics, miRnome, copy number variation, next gen sequencing). He reviewed the emerging opportunities for the omics technologies in this area. Particularly, he showed that microarray expression analysis of peripheral blood mononuclear cells (PBMCs) could serve as tissue surrogate to reflect changes in the lung vascular wall and the right ventricle[37,38] He pointed out that in neoplastic disorders, the cancer cell line encyclopedia enables predictive remodeling of anticancer drug sensitivity. Similar use of gene expression analysis should allow us to detect reproducible signatures, to accurately classify predefined classes and/or to define new molecular phenotypes of P(A) H linked with preferred responsiveness to specific drugs. Along this line, Harrison Farber showed that gene expression and cytokine profiles are clearly distinct in PBMCs isolated from limited systemic sclerosis (lSSc) with PAH (ISSc-PAH) as compared to ISSC without PAH (ISSc-no PAH). Interferon gamma related genes, especially mannose receptor C type 1 (MRC1) expression was increased exclusively in activated monocytes of lSSc-PAH patients and correlated strongly with PAP and increased mortality.
Allen Johnson provided in-depth knowledge on different computed tomography (CT) modalities, ranging from dynamic contrast-enhanced CT to micro CT to micro-SPECT, in order to discriminate between various types of vascular abnormalities in PH. Lan Zhao explored the utility of fluorodeoxyglucose positron emission tomography (FDG-PET) in experimental models of PAH in response to DCA and Imatinib. Increased FDG uptake observed in PAH rats was strongly reduced by both DCA and Imatinib treatment. Moreover, Martin Wilkins provided evidence that this technology may also be employed in human PAH. This may open a new avenue for direct visualization of anti-proliferative treatment effects in patients with PAH.
Further novel approaches include the analysis of microRNA in plasma samples and the use of patient derived iPS cells for individual ex vivo drug testing. Utilization of one or several of these approaches could lead to less invasive screening, better understanding of individual pathogenesis and improved personalized treatment, but we are at best at the beginning of solving this challenge.
BEYOND PH-VASCULAR ABNORMALITIES IN COPD
COPD is one of the most common causes of death worldwide. Alterations in pulmonary vessel structure and function are highly prevalent in patients with COPD. Importantly, more than one third of patients with COPD also have PH, shown to be inversely associated with survival in these patients. Changes in the pulmonary circulation have been identified at initial disease stages, providing new insight into their pathogenesis.[39] Joan Barbera focused on this aspect and summarized cellular and molecular mechanisms of COPD-associated PH (Fig. 5). He showed that intimal hyperplasia and endothelial dysfunction of pulmonary arteries represent characteristic features of COPD, present even at early disease stages and in smokers without airflow obstruction. Accumulation of inflammatory cells, release of growth factors and endothelial dysfunction underlie vessel remodeling in smokers. He highlighted that bone marrow-derived progenitor cells feed poorly differentiated SMCs in the hyperplasic arterial intima under these conditions. These pathologic morphologies could be imaged to obtain clinically relevant information in patients with COPD. Using imaging and clinical data from the COPD gene study, George Washko reported that quantitative CT based assessment of intraparenchymal vessel dilatation, vascular pruning and reduced vessel number are associated with clinical manifestations of smoking related lung diseases.
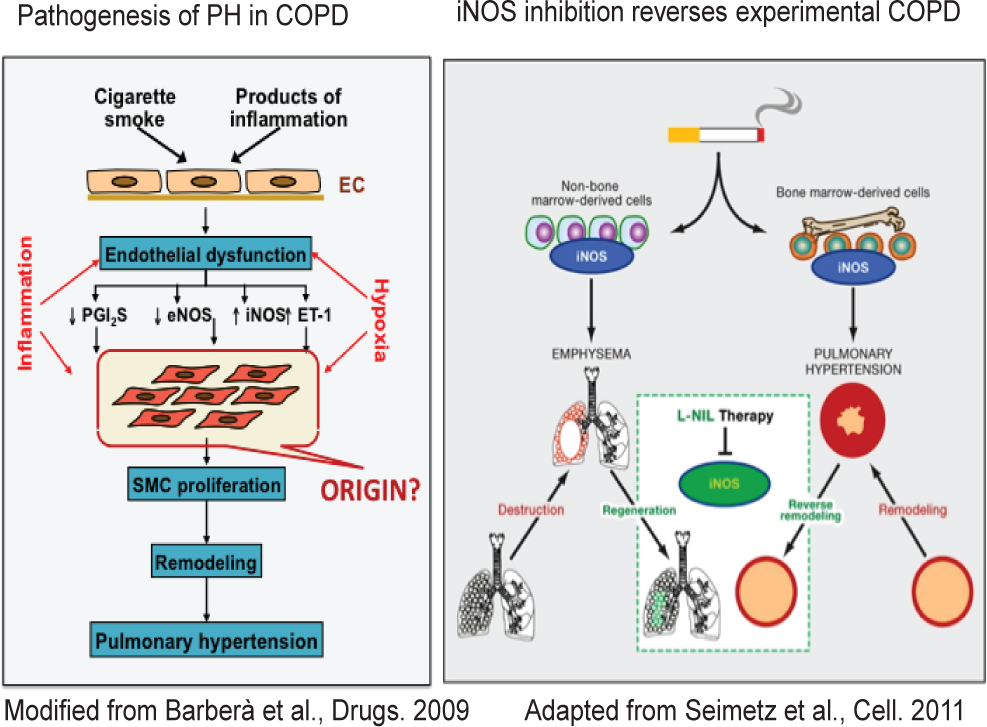
(Left) Cellular and molecular mechanisms of COPD - associated PH. (Right) Novel therapeutic strategies: Inducible nitric oxide synthase (iNOS) inhibition reversed COPD - associated PH and emphysema.
Progress in the understanding of the pathobiology of PH associated with COPD may provide the basis for a new therapeutic approach. In this respect, Norbert Weissmann's group demonstrated that mice chronically exposed to tobacco smoke have pulmonary vascular remodeling and PH preceding emphysema formation.[40] They provided strong evidence for a causative role of iNOS in both PH and emphysema formation, via contribution of both bone marrow-derived as well as intrapulmonary stem cells. Importantly, treatment with iNOS inhibitor both prevented and reversed structural and functional alterations of both the lung vasculature and the alveolar compartment. The safety and efficacy of iNOS-targeted therapy in COPD-associated pulmonary hypertension warrants further investigation in randomized clinical trials.
CONCLUSIONS
Where are we in PH research in 2012? Beyond doubt, we made major progress to identify important cellular and molecular players of PH and most importantly to profit from this knowledge for shaping new therapies. However, we are still far away from a comprehensive understanding of this deadly disease. This is true for the proliferative abnormalities of the pulmonary vasculature and is even more true for the pathogenetic sequelae underlying right ventricular hypertrophy and failure. Personalized therapy (optimally balancing risks and benefits of different treatment strategies for individual patients) is not more than a faint loom at the horizon. There is substantial doubt that we will ever find a “metabaloVasodilApopStat-D” (Curesitall), alluding to Stephen Archer's persiflage. But, we are and will stay dedicated to unravel step by step molecular mechanisms contributing to PH and profit from such findings to shape novel therapeutic concepts, in the best sense of an interdisciplinary and translational approach.