
Abstract
Survival rates for patients with idiopathic pulmonary arterial hypertension (IPAH) have improved with the introduction of PAH-specific therapies. However, the time between patient-reported onset of symptoms and a definitive diagnosis of IPAH is consistently delayed. We conducted a retrospective, multi-center, descriptive investigation in order to (a) understand what factors contribute to persistent diagnostic delays, and (b) examine the time from initial symptom onset to a definitive diagnosis of IPAH. Between January 2007 and December 2008, we enrolled consecutively diagnosed adults with IPAH from four tertiary referral centers in Australia. Screening of patient records and “one-on-one” interviews were used to determine the time from patient-described initial symptoms to a diagnosis of IPAH, confirmed by right heart catheterization (RHC). Thirty-two participants (69% female) were studied. Mean age at symptom onset was 56 ± 16.4 years and 96% reported exertional dyspnea. Mean time from symptom onset to diagnosis was 47 ± 34 months with patients subsequently aged 60 ± 17.3 years. Patients reported 5.3 ± 3.8 GP visits and 3.0 ± 2.1 specialist reviews before being seen at a pulmonary hypertension (PH) center. Advanced age, number of general practitioner (GP) visits, heart rate, and systolic blood pressure at the time of diagnosis were significantly associated with the observed delay. We found a significant delay of 3.9 years from symptom onset to a diagnosis of IPAH in Australia. Exertional dyspnea is the most common presenting symptom. Current practice within Australia does not appear to have the specific capacity for timely, multi-factorial evaluation of breathlessness and potential IPAH.
Idiopathic pulmonary arterial hypertension (IPAH) is a fatal disease that, in recent years, has witnessed an intense focus on preclinical and clinical research. In the last decade, 18 randomized, placebo-controlled clinical trials have been completed, culminating in the availability of seven PAH-specific therapeutic options.[1–18] While survival in treated prevalent (existing) cases and clinically stable patients enrolled in randomized clinical trials has improved dramatically, incident case-fatality has only marginally improved.[19,20] IPAH is a disease that can be ultimately diagnosed only by right heart catheterization (RHC) and is defined by a mean pulmonary artery pressure (mPAP) < 25 mmHg, with a normal pulmonary artery wedge pressure (PAWP) and an elevation in pulmonary vascular resistance (PVR) of more than three Wood units.[21] RHC usually follows a comprehensive clinical evaluation to exclude other causes of pulmonary hypertension (PH) that have identifiable associations with elevated pulmonary pressures (e.g., left heart disease, lung diseases, thromboembolic disease, congenital heart disease and connective tissue disease).
The most commonly described symptoms reported by patients diagnosed with IPAH are shortness of breath and fatigue.[22–24] Significantly, the time between patient recognition/reporting of symptoms and a definitive diagnosis of IPAH is consistently delayed, regardless of socioeconomic, cultural and geographical profile of the reporting country.[22,24–26] Historically (prior to the introduction of PAH-specific therapies in the 1990s), a delay of approximately two years to diagnosis was reported.[24,27] Since that time, the reported epidemiological data relating to PH has evolved, as more and more patients are identified.[24,25,28] Unfortunately, two recent studies from Europe[29] and the United States[30] evaluating the burden of PAH have confirmed that a significant and potentially adverse delay in diagnosis persists, which may impact on prognosis even with therapy.
In order to understand what factors contribute to this delay, we conducted a retrospective, multi-center descriptive investigation (the DELAY Study) of patients' journeys from symptom onset to definitive diagnosis of IPAH.
MATERIALS AND METHODS
Study setting
In Australia, current restrictions mean that PAH-specific therapy can only be prescribed from a designated center of excellence. Designated centers, of which there are 49, are health facilities that have met the criteria set by the Department of Health and Aging for referral, diagnosis, and management of PH. Centers of Excellence are publicized to physicians by Medicare Australia.[31]
Between January 2007 and December 2008, we enrolled consecutively diagnosed IPAH patients from four geographically diverse centers of excellence. These centers, the Royal Perth Hospital, Perth, Western Australia; The Alfred Hospital, Melbourne, Victoria; The Prince Charles Hospital, Brisbane, Queensland; and St. Vincent's Hospital, Sydney, New South Wales, manage referrals from city, rural and remote areas (Perth and Queensland) and large metropolitan cities in addition to some rural referrals (Sydney and Melbourne). The study was approved in each center by the relevant Human Research Ethics Committees (HREC).
Participants
All patients underwent formal screening assessments as per guidelines published by the European Society of Cardiology (ESC)[32] and the American College of Chest Physicians (ACCP).[33–35] This included a physical examination, Chest X-ray, Doppler echocardiogram, pulmonary function tests (to rule out significant lung disease), a ventilation-perfusion scan (to rule out chronic thrombo-embolic PH) and exercise capacity, as measured by a 6-Minute Walk Test (6 MWT).
Consecutive patients were eligible for inclusion if they had a diagnosis of IPAH by RHC at a center of excellence during the period from January 2 007 to December 2008. IPAH was defined by an RHC procedure with an mPAP < 25 mmHg at rest, a PAWP below 15 mmHg and a PVR < 3 Wood units with no other associated cause identified. Patients > 18 years of age and/or those with any other etiology of PH were excluded. All subjects gave written informed consent.
During the study period, 160 patients were screened across the four participating centers and 40 (25%) with a diagnosis of PAH were identified. However, eight (20%) were removed from the final analysis, seven were ineligible due to subsequent identification of an associated cause of their PAH and one did not present for the interview.
Study data
We examined the time from patient-described initial symptoms to an RHC diagnosis of IPAH. Data were also collected on the time from first medical contact to RHC, time from RHC to initiation of PAH-specific therapy, number of GP and specialist reviews prior to RHC and the number of alternative diagnoses prior to IPAH. Patients then participated in a “one-on-one” quantitative and qualitative standardized interview by an interviewer blinded to the medical history of the patient. Each patient's medical record was then retrospectively interrogated to document patient characteristics at the time of IPAH diagnosis. All data were validated by a subsequent discussion with the specialist center physician (Fig. 1). Each patient was prospectively followed to a censor date of October 1, 2011 to evaluate case-fatality and any potential relationship to delayed diagnosis.
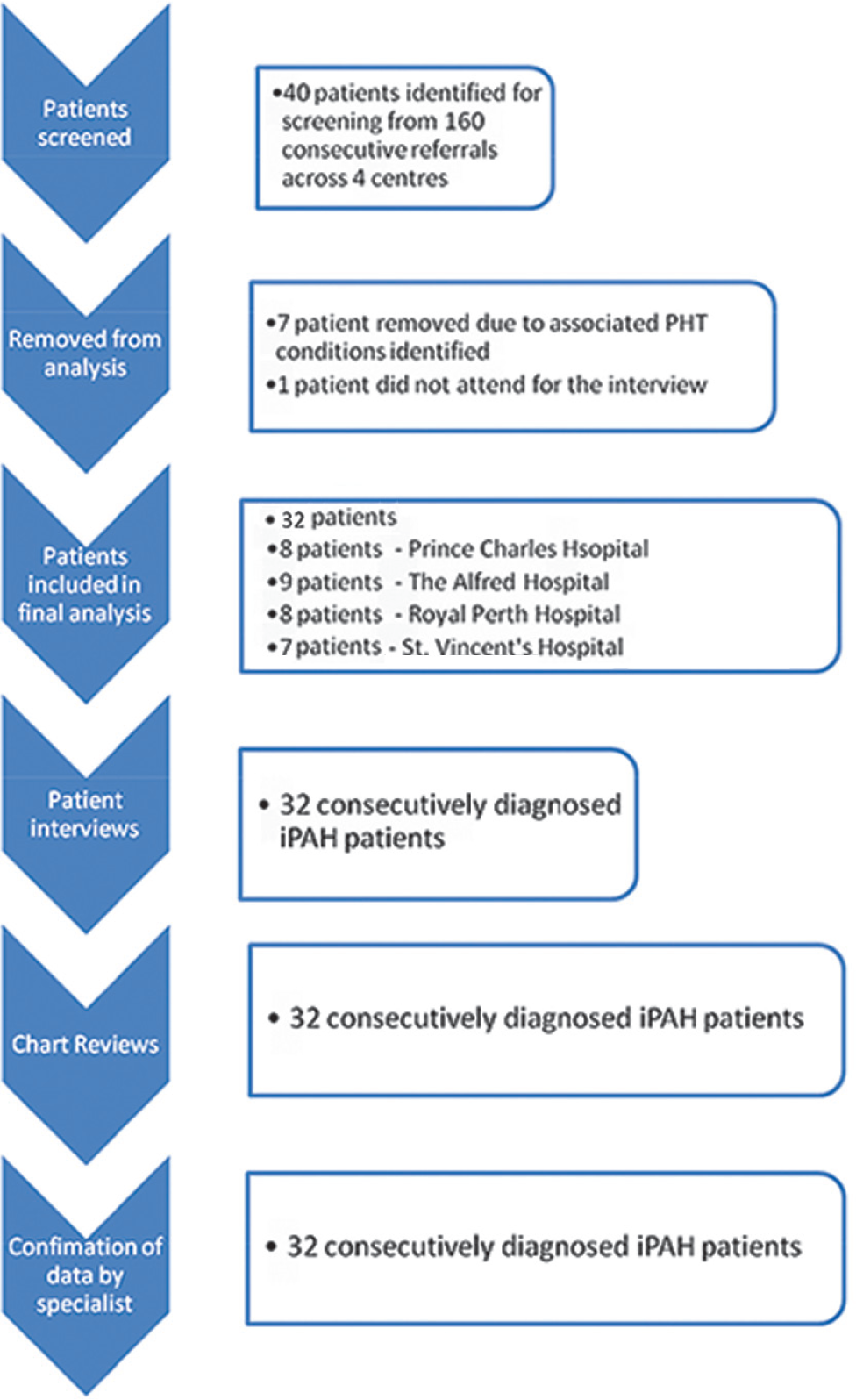
Study schema.
Data analyses
All data is presented using descriptive statistics showing mean (± standard deviation) and medians (± 25th and 75th interquartile ranges [IQR]). A Wilcoxon sum test was used for nonparametric hypothesis testing comparison between males and females for (a) time to diagnosis, (b) time from first medical contact to diagnosis, (c) age at symptom onset, and (d) number of GP visits. Logistic step-wise multivariate regression analyses were used to review possible correlates with time to diagnosis. We included the following covariates in our modeling: Age at symptom onset, gender, systolic blood pressure (SBP), heart rate (HR), PVR, mRAP, mPAP, cardiac index (CI), number of specialist visits and the number of GP visits. Finally, we employed a log and square root transformation in our validation of the model for age at diagnosis, HR, SBP and number of GP visits to account for non-normally distributed variables.
RESULTS
Patient demographics
Table 1 summarizes the demographic and clinical profile of the 32 study participants (69% female) at diagnosis. Overall, mean age was 56 ± 16 years at the time of symptom onset and 59 ± 17 years at the time of diagnosis by RHC. At symptom onset, males were older than females (mean age 58 ± 17.74 vs. 53 ± 15.51 years). The hemodynamic and functional profile of participants was consistent with a diagnosis of IPAH, with an mPAP of 44 ± 13 mmHg, accompanied by markedly impaired exercise tolerance as measured by the 6 MWT (mean 290 ± 142 m). The most commonly reported symptom (96% of patients) was exertional dyspnea, often relatively subtle (e.g., walking up stairs, not being able to run as “far as usual,” walking up hills, or not being able to keep up with peers); equivalent to World Health Organization (WHO) Functional Class II at symptom onset. Ninety-five percent of patients described their symptoms as “a mild limitation” when they were first recognized (WHO Functional Class II) and 5% described “marked limitation” as the first recognition of symptoms being present (WHO Functional Class III). In contrast, 94% of patients were in WHO Functional Class III and 6% in WHO Functional Class IV at the time of diagnosis (Fig. 2).
Patient demographics
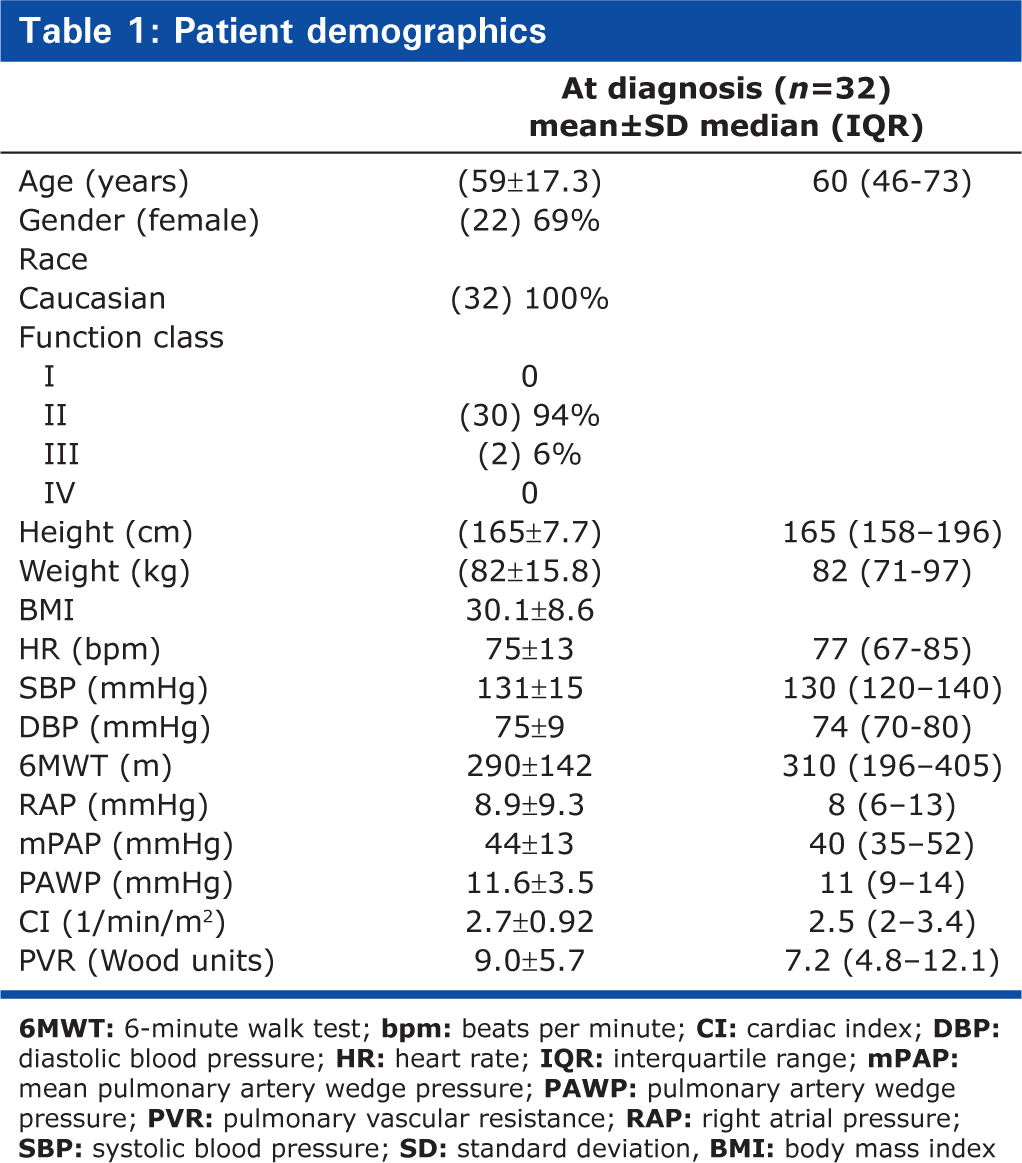
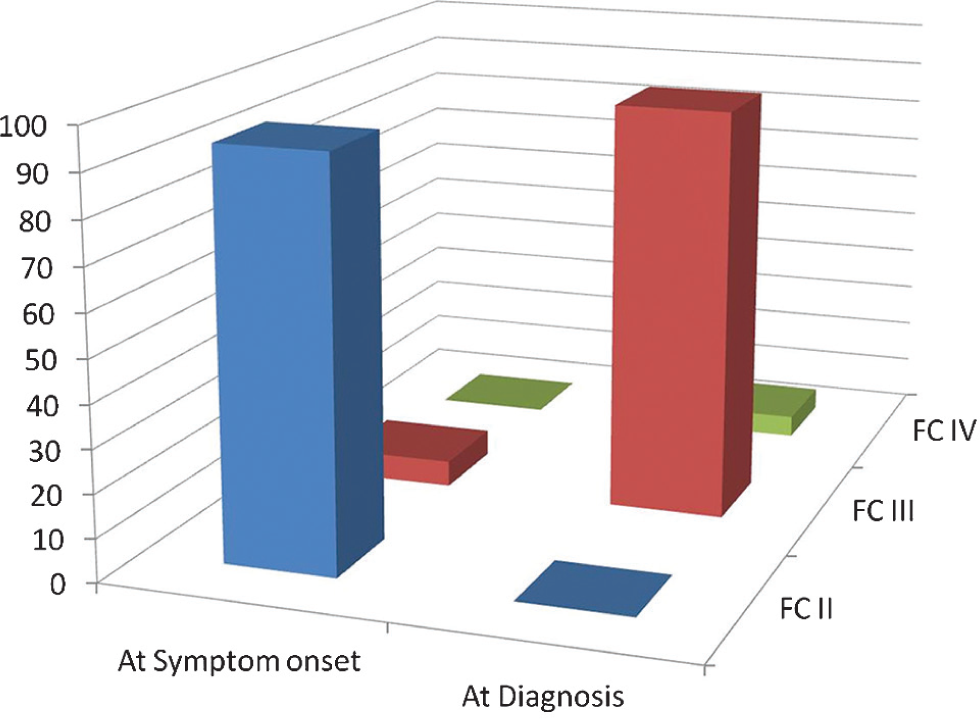
WHO Functional Class at symptom onset and at definitive diagnosis. FC, Word Health Organization modified functional class. At symptom onset, 95% of patients were classified with WHO FC II disease and 5% with WHO FC III. At diagnosis, the distribution between FC II, III and IV in the same patients was 0%, 94% and 6%, respectively.
Time to (delayed) diagnosis
The mean time from symptom onset to diagnosis at RHC was 47 ± 34 (Median 44, IQR: 21-65) months. Mean time to RHC diagnosis from first medical contact was 36.5 ± 34.7 (Median 30, IQR: 10-58) months.
Diagnostic journey
On average, patients received two “alternative diagnoses” prior to the RHC confirmed diagnosis of IPAH. Thirty-three percent of patients (
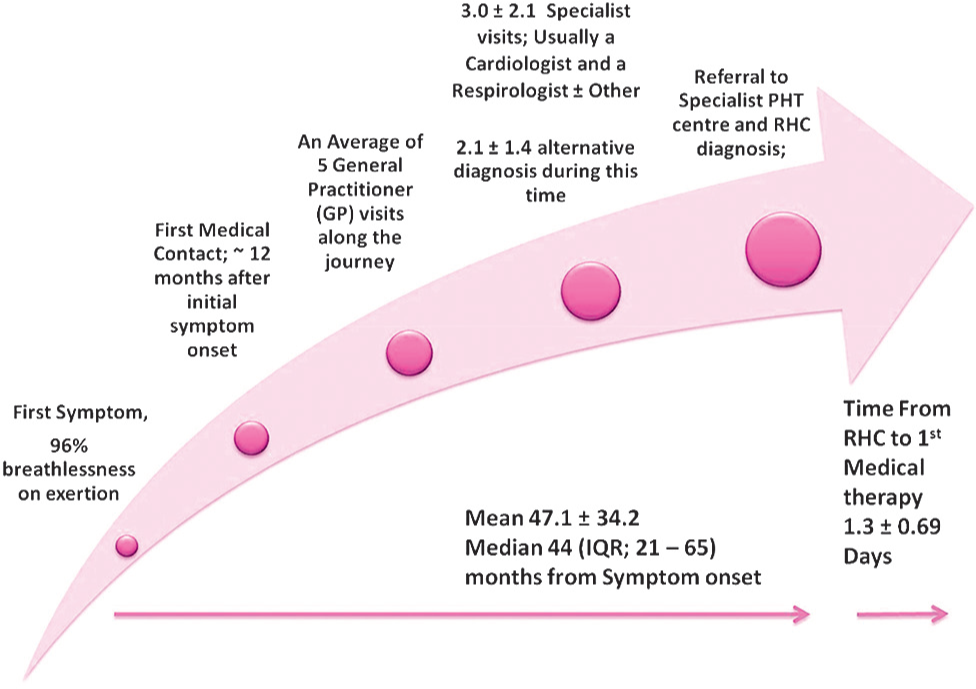
Gender difference throughout diagnostic journey. Differences between males and females (means ± 95% CI) with regard to age at symptom onset, Time to diagnosis, number of GP visits and time from first medical contact to diagnosis.
On an adjusted basis, increases by 10 beats per minute in HR shortened the time to diagnosis by 0.674 months (CI: −1.283–0.065;
Case-fatality
In the three years following the original interviews of our cohort, three patients (9.4%) died. Due to the very small numbers, we did not perform an analysis of factors influencing survival.
DISCUSSION
In this unique Australian study, we identified a significant delay in the time to referral and definitive diagnosis of IPAH in patients managed by centers of PH excellence. We note an average delay from symptom onset to RHC of 47 ± 34 months (3.9 years). There is a significant period of approximately 12 months from initial symptom onset, as recognized by the patient and subsequent presentation to a medical practitioner. We have documented a significant deterioration in functional class during this time and noted a correlation between the delay in diagnosis and primary care parameters such as the number of primary care visits, HR, SBP and age.
It has been 30 years since the National Institutes of Health (NIH) commenced the first registry on the then called “primary pulmonary hypertension or PPH.”[24] In that registry, data was collected over a five-year period, culminating in 187 PPH patients entered from 32 centers across the United States. The cohort contained a 1.7:1 ratio of females to males who were classified in either New York Heart Association (NYHA) Functional Class III or IV in 75% of cases with a mean age of 36.4 years. The average time to diagnosis from symptom onset was reported as 2.03 ± 4.9 years and was similar for both sexes. Although 90% of this cohort had symptoms for less than three years, 10% reported symptoms of up to 20 years in duration. A similar contemporaneous study of 91 PPH patients referred for heart lung transplantation (aged 29.8 ± 7.9 years at PPH diagnosis) had symptom duration of 65.9 ± 47.4 months, while survival from diagnosis was reported as 42.9 ± 42.6 months, giving a mean “lead time” from symptom to diagnosis of 23 months.[27] These two US-based studies in the same year suggest a homogeneous time from symptom onset to diagnosis of 24 months at the time of reporting. In the current era, we note a very different age demographic in our patient population (60 ± 17.3 years) with much longer disease duration prior to diagnosis (47 ± 34 months).
Since the late 1980s, two large registries have published data pertaining to the delayed diagnosis of PH, including the French National[25] and REVEAL (North America) registries.[30] In 2006, the French registry reported a delay of 27 months (2.3 years) from reported symptom onset to diagnosis.[25] The REVEAL registry reported a delay of 35 months (2.8 years) from a heterogeneous cohort that included 1,166 IPAH patients enrolled between March 2006 and September 2007.[30] The French data have been replicated in Europe in a “burden of disease” study from Germany. That group reported a mean delay of 2.3 ± 3.7 years in a similarly aged population (mean age 55 years). We report a delay of 10 to 12 months longer than the REVEAL data (47 months). The average age of patients enrolled in the REVEAL, French and German cohorts were four to six years younger than our average age at diagnosis and may have implications on gradual increase in “delays in diagnosis” as the population demographic changes from the reports from the NIH[24] and Stanford registries[27] of the 1980s. The increased age of affected individuals may account for some of the delay in recognition as clinicians transition from expectations of a young (30s) female and adapt to the possibility of IPAH being present in men and women of all ages of the population, with the average IPAH patient now in their fifth decade of life presenting with shortness of breath.[19,30,36,37]
Patients reported, on an average, having five visits with their GP and two specialist visits before being referred to a PH center. We did not quantify the exact time this contributed to the delay. We did quantify the time delay attributable to the patient's lack of presentation to a medical practitioner, which equated to delayed presentation for approximately 12 months. This implies that the current structure of healthcare provision in Australia for PH, delays patient diagnosis of IPAH, by approximately three years within the combination of primary and specialty care delivery. Interestingly, this figure correlates well with other previously reported figures of delays in diagnosis. It would be reasonable to suggest the current differences in reports of delay may under-represent the true delay, if specific questions were asked directly on first symptom recognition. We postulate that the current structure of breathlessness management does not expedite definitive patient care in subspecialty conditions, such as PAH. Only one study to date has looked at the burden of disease in PH.[29] In that study, the authors expose the relatively high numbers of healthcare resources utilized by patients with IPAH. Breathlessness, reported in 96% of our cohort, can be a chronic and certainly complex phenomenon which has been shown to create a significant burden to those who suffer from it, carers and the healthcare resources.[38] Moreover, it accounts for more than 900,000 GP presentations in Australia.[39] A multidisciplinary approach to breathlessness management coupled with a more expedited process leading to simple RHC may indeed shorten the time to diagnosis for IPAH and many other diseases where breathlessness of unknown causes is the primary symptom to be addressed.
Age, HR and SBP were correlates of delayed diagnosis and the time to diagnosis in men was 17 months longer than that observed in women (although not statistically significant). It is difficult to speculate as to why HR and SBP were positive correlates; however, it would appear intuitive that over an extended period, more visits to a GP for unresolved issues (e.g., hypertension) are likely to occur. With regard to the delayed diagnosis in males versus females, there is some evidence to suggest that older males are more likely to defer treatment for cardiovascular conditions than females.[40,41] However, whether this factor applies to PH remains speculative and requires further investigation.
In the present study, we noted a shift (deterioration) of one functional class during the delay period. Functional class has been correlated to survival in many studies of IPAH and has been validated as an independent predictor of survival.[42–44] Recently, Humbert and colleagues demonstrated the impact of functional class on survival in incident IPAH cases.[19] In that study, the authors report a 40% survival at 24 months for those patients in Functional Class IV compared with a 90% survival at 24 months for those patients in Functional Class II. During the time of delay in our study, we noted a substantial decline in functional class, which, based on the observations by Humbert, et al., may have a negative impact on survival, increasing mortality up to 20% over a 24 month period.
There are a number of limitations in the present study that warrant mention. First, this was a small patient cohort, despite being large in the overall context of the likely pool of IPAH patients in Australia and may not accurately reflect contemporary patient journeys with the same disease. We also relied on patient description of the onset of symptoms and this may lead to bias in over-or underestimating the time of symptom onset. All our patients were diagnosed at RHC after excluding other forms of PH, so we are satisfied that the time of diagnosis is correct. Because this was a small cohort, we cannot draw any firm conclusions on any potential relationship between the two, or attribute low mortality to increased utility of PAH-specific drugs. Therefore, further studies are needed to determine the mortality impact of timely diagnosis in this patient population.
In conclusion, in this small but unique study, we report a significant delay of almost four years from symptom onset to a diagnosis of IPAH. Further, the delay appears to have three components: (1) patient-driven delay in presentation; (2) GP delays in referral; (3) specialist delays over multiple reviews prior to referral/presentation at a PH center. These represent natural points for health service interventions to provide more rapid and definitive diagnosis of IPAH. During that delay, patients described a deterioration of functional class, which, based on observations from previous studies, may have an impact on mortality. Breathlessness on some form of exercise is the most commonly reported symptom in patients diagnosed with PH. Current practice within Australia does not appear to have the specific capacity for timely, multi-factorial evaluation of breathlessness.