
Abstract

The search for effective treatments for pulmonary hypertension (PH) has been frustrating. The very first drug for idiopathic pulmonary arterial hypertension (PAH), epoprostenol, was approved in 1995.[1] And while progress has been made, a look at where we were, where we are, and where we need to be suggests we are far from accomplishing our goals.[2] We have identified three classes of drugs that improve symptoms, but we still do not have drugs that modify the disease process or protect patients from developing progressive pulmonary vascular disease.[3] Perhaps this is because the developed drugs were studied due to their vasodilator properties, while scientific research has now demonstrated that cellular proliferation, inflammation, and thrombosis are the dominant underlying pathobiologic processes, with chronic pulmonary vasoconstriction playing a relatively minor role.[4]
In a recent editorial, Dr. Joseph Loscalzo pointed to the continuing low level of drugs approved by the FDA over the past 11 years in the face of an increasing demand for personalized cardiovascular drug development.[5] His call for a new paradigm has been echoed by academics, regulatory agencies, and the pharmaceutical industry. However, with limited patients and the high cost of drug development, it has become obvious that we have to find ways to identify promising drugs more quickly, with trials that require smaller numbers of patients, and shortened times to approval if we expect the pharmaceutical industry to continue to invest in treatments for this disease.[6] In return, our industry partners must be willing to embrace a paradigm that requires data sharing and collaboration.[7]
To address this challenge, the Pulmonary Hypertension Academic Research Consortium (PHARC) was created as a forum to openly discuss strategies for clinical trials in PH that would benefit all of the stakeholders. The consortium initially included academics with an interest in pulmonary vascular disease, regulatory authorities from the United States, Canada, and Mexico, members of pharmaceutical companies with approved or developing drugs for PH, and observers from medical societies from North America (Appendix 1 on page 267). The goals of the consortium were to establish consensus clinical endpoint definitions for future clinical trials, advance the conduct of clinical research in the field, identify modern strategies for clinical trials, and provide guidance to the pharmaceutical industry to allow them to better identify and develop treatments with the most promise. In the spring of 2012, the first meeting of the PHARC was held in Bethesda, Maryland, USA.
The PHARC established five working groups and a pediatric advisory committee to focus on specific areas for discussion. Working Group 1 was charged with reviewing the collective experience from previous clinical trials to guide future studies. One of their topics was the definition of the clinical phenotypes of PH for entry into clinical trials. It is not widely appreciated, but clinical trials and the regulatory approval of drugs for PH follow a clinical classification scheme that was first proposed at an international World Health Organization (WHO) sponsored symposium in 1998.[8] The classification system, however, was never intended to guide therapies or dictate clinical trial design. It was developed as a guide for physicians to assist in making the clinical diagnosis of PH. As a result, the majority of patients with PH have been ignored in registration trials, as there is not a single approved treatment for patients with PH associated with heart or lung diseases. Whether this is correct, or even appropriate, remains untested. If, for example, we were to define severe PH based on the level of elevation of mean pulmonary artery pressure, one could defend including patients from Category 1 (PAH), Category 2 (left heart disease), and Category 4 (chronic thromboembolic disease) in a single clinical trial (Fig. 1). This is one area that urgently needs to be explored because there remains an unmet medical need for the majority of patients suffering from PH.
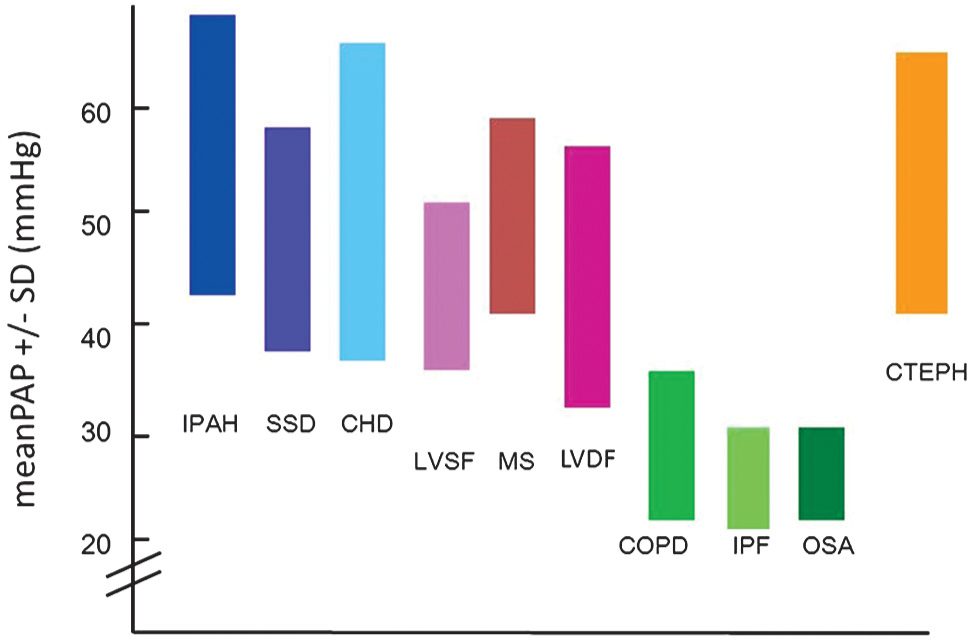
The mean pulmonary arterial pressure range of one standard deviation from the average is shown for patients with pulmonary hypertension from several etiologies. The values are derived from published registries, case series, and clinical trials. Note the similarity from each of the etiologies with the exception of the lung diseases. Legend: IPAH = idiopathic pulmonary arterial hypertension; SSD = scleroderma spectrum of diseases; CHD = congenital heart disease; LVSF = left ventricular systolic failure; MS = mitral stenosis; LVDF = left ventricular diastolic failure; COPD = chronic obstructive pulmonary disease; IPF = idiopathic pulmonary fibrosis; OSA = obstructive sleep apnea; CTEPH = chronic thromboembolic pulmonary hypertension.
Another consideration is whether there is a need to categorize patients based on the associated diseases and study or analyze them separately as opposed to bunching these groups together under WHO Group 1 PAH. The natural history of the PAH subgroups is very different.[9] The early epoprostenol studies conducted separate trials for idiopathic PAH and PH secondary to connective tissue disease with different results.[1,10] While pooling all patients with PAH into a single trial eases patient recruitment, it may provide misleading information about effective therapies. In addition, there appears to be a wide spectrum of responsiveness to drugs from patient to patient which has not been addressed. If alternative trial designs or endpoints could be used, then a strategy of studying or analyzing the PH subgroups separately could be adopted. Given the differences in the disease within these heterogeneous subgroups, it is important to clearly define the phenotype of these populations both for the inclusion in clinical trials as well as for reporting purposes.
Working Group 2 was charged with proposing new or more clinically relevant clinical endpoints. The 6-Minute Walk Distance (6 MWD) test has been the primary endpoint in the majority of registration trials for PH. As is tradition, because it was selected as the primary endpoint in the first successful registration trial with intravenous epoprostenol for primary PH, it has been used as the primary endpoint in registration trials for PH ever since. The 6 MWD has been recently subjected to intense criticism because it does not appear to reflect the wellness or lack of wellness from the treatment in patients in these trials.[11] Composite endpoints such as time to clinical worsening or time to clinical improvement may be more appropriate.[12] The testing of surrogate endpoints and identification of important biomarkers is also needed.[13]
Working Group 3 reviewed the current science of clinical trials and how modernizing clinical trial designs may be advantageous when studying PH. These include adaptive trial designs Bayesian methodology, adjusting for combination therapies, and prespecifying subgroup analyses.[14] A living example of how this could make a difference can be extrapolated from the experience in using high doses of calcium channel blockers in vasoreactive patients with PH. If calcium channel blockers were to be tested with the current approach, it is possible that the dramatic efficacy in the small subset of vasoreactive patients would get diluted in a traditional trial resulting in the conclusion that calcium channel blockers are ineffective. Conversely, if the vasoreactive patients had a large enough improvement in the primary endpoint, it might be possible that calcium channel blockers would receive regulatory approval as a treatment for PH in all patients. Either way the results would be erroneous. However, with our current knowledge about this disease, the appropriate clinical trial design would be to prespecify vasoreactivity as a biomarker by which enrollment into the trial would be stratified. Given the dramatic efficacy that calcium channel blockers have in this group, it is quite probable that a clinical trial utilizing less than 100 patients would be sufficient to identify those patients for whom this drug should receive regulatory approval.
Working Group 4 reviewed current translational research and the classes of new medications and molecular targets that are likely to be relevant in the next several decades. Given the complexity of the molecular pathways that have been identified as involved in human PH,[15,16] it is highly unlikely that a single drug will be effective in the majority of patients. In addition, as drugs are targeted to specific molecular pathways identified by animal models, we embark on novel classes of drugs that are potentially harmful as well as helpful. One important example has been the recent experience with dasatinib,[17] a tyrosine kinase inhibitor that was discovered to cause PH at the same time that an international clinical trial was investigating imatinib,[18] a related tyrosine kinase inhibitor, as a treatment of PH.
Working Group 5 addressed the essential academic standards in the conduct of clinical trials, which need to be adhered to as we move forward. The outcome of a candid and open discussion of these issues was a consensus that a formal international pulmonary hypertension clinical trial consortium be created. Key to the success of this initiative will be input from the regulatory authorities regarding acceptable clinical trial designs and the possibility of provisional approvals to allow subsequent trials to better identify the risks and benefits of a therapy. We must create an environment that the industry perceives as potentially profitable if we expect their continued investment in this disease. At the same time, the pharmaceutical industry must drop their proprietary mentality and allow openness in the analysis of these trials by giving unrestricted access of the database to an academic steering committee.[7] We need to be allowed to find out in whom the drugs work and in whom they do not. Academic physicians similarly must address the influence of industry that has resulted in a culture of financial blindness, by limiting the physicians who receive monetary compensation from these companies from having undue influenced.[19] Once and for all, the era of ghostwriting manuscripts should be declared gone forever.[20]
Finally, the Pediatric Advisory Committee was charged with identifying those issues relevant to the pediatric population, including neonatal and adolescent populations, and how they can best be addressed and anticipated in future studies. Since PH is a disease affecting all ages,[21] it is no longer acceptable to ignore this important group from clinical research. The PHARC rejects the notion that registration trials be conducted on adults, with consideration of the pediatric patients only after a treatment has won approval. Proof of concept or Phase II trials in pediatric patients can and should be conducted while clinical trials for adults are ongoing.
Summary reports from each of these working groups are being published in this issue of Pulmonary Circulation for all to review. It is the hopeful and ambitious goal of the PHARC that we can establish ourselves as a role model for the international medical community in the conduct of clinical research toward developing future therapies.