
Abstract
Acute massive pulmonary embolism results in a rise in pulmonary vascular resistance and acute right ventricular dilation and systolic dysfunction. In unstable patients who cannot safely undergo computed tomography, echocardiography can often provide direct and indirect evidence of pulmonary embolism. We present a case in which clinical and echocardiographic evidence provided sufficient evidence of PE to initiate treatment. Dynamic improvement in hemodynamics and echocardiographic parameters of RV function indicated successful treatment.
Three-month mortality in hemodynamically unstable patients with acute pulmonary embolism (PE) approaches 60%, compared to approximately 15% in hemodynamically stable patients and evidence of right ventricular (RV) hypokinesis on echocardiography doubles the risk of death at 30 days.[1] In stable patients with high clinical suspicion of PE, computed tomography is the diagnostic test of choice; ventilation/perfusion imaging may be pursued in patients with a contraindication to iodinated contrast. In unstable, critically ill patients CT remains the test of choice if it can be performed without delay, but transthoracic or transesophageal echocardiography should be performed if CT is not immediately available or would result in additional risk.[2] In these patients, the combination of high clinical suspicion for PE and echocardiographic findings suggestive of PE may be sufficient to initiate treatment.[3]
This case presents a patient in whom diagnostic options were limited and echocardiography provided strong circumstantial evidence for a massive acute PE. The case also demonstrates the afterload dependence of RV function as measured by quantitative echocardiographic parameters of RV function.
CASE REPORT
A 57-year-old woman with a history of Non-Hodgkin's Lymphoma thought to be in remission presented with the acute onset of dyspnea, fever, and hemoptysis. Admission chest imaging and bronchoscopy were diagnostic for diffuse alveolar hemorrhage secondary to adenovirus pneumonia. An admission transthoracic echocardiogram was normal, including normal biventricular chamber size and function. Chemical thromboembolic prophylaxis was held on hospital day (HD) four given her life-threatening pulmonary bleeding; sequential compression devices were applied until HD 12, when prophylactic heparin was resumed. With ventilatory support, parenteral steroids and eventual diuresis, the patient's pulmonary status improved over the subsequent two weeks.
On intensive care unit Day 15 (HD 17), the patient developed the acute onset of rapidly increasing vasopressor requirements, refractory hypoxia, and tachycardia despite improvement in her airway pressures and the appearance of her chest imaging. A repeat bedside transthoracic echocardiogram revealed normal left ventricular function, but severe RV dilation with qualitative and quantitative evidence of RV hypokinesis (Fig. 1). Chest imaging to confirm the presence of a pulmonary embolism could not be obtained given the patient's reduced glomerular filtration rate and hemodynamic instability.
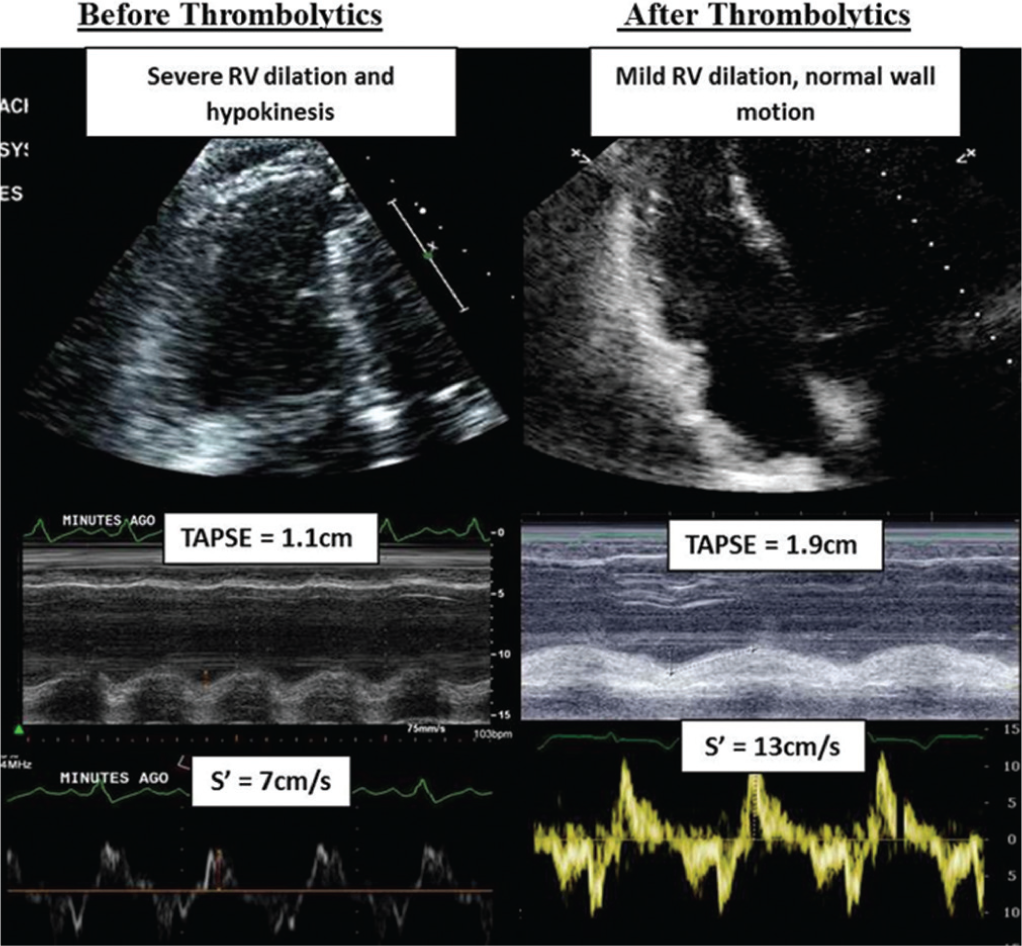
Echocardiographic assessment of right ventricular (RV) function before and after thrombolytics. These images demonstrate qualitative and quantitative improvement in RV size and systolic function. TAPSE = tricuspid annular plane systolic excursion; s' = myocardial velocity of the lateral tricuspid annulus.
Given the acute development of signs, symptoms and imaging changes consistent with an acute, massive pulmonary embolism, the treatment team administered tissue plasminogen activator 10 mg given as a bolus, followed by 90 mg given as an infusion over the subsequent two hours. At the completion of the infusion, the patient's vasopressor requirement decreased from norepinephrine 20 mcg/min and vasopressin 0.04 units/min to being off all vasopressors, her oxygen requirement decreased from a fraction of inspired oxygen of 100% to 40%, and her heart rate decreased from 130 to 90 beats/min. Repeat transthoracic echocardiogram the following day showed mild RV dilation, resolution of RV dysfunction (Fig. 1), and a decrease in estimated pulmonary vascular resistance.
DISCUSSION
Echocardiography has limited sensitivity and specificity for the diagnosis of acute PE;[4] however, in many patients with a pulmonary embolism large enough to cause hemodynamic instability, direct or indirect evidence will be present. Direct visualization of a saddle embolus may be seen, particularly on transesophageal echocardiography. Indirect evidence of PE includes right ventricular dilation and hypokinesis, tricuspid regurgitation, elevated pulmonary artery pressure on Doppler, septal flattening, and dilation of the inferior vena cava with loss of respiratory variation. The assessment of right ventricular function in patients with suspected PE should include both qualitative visual evaluation and quantitative parameters. Quantitative parameters such as the tricuspid annular plane systolic excursion (TAPSE) and myocardial velocity of the lateral tricuspid annulus (S') are easy to acquire and reproducible with the limitation that both these measures extrapolate basal RV function to assume global RV function. Fractional area change (FAC) is a more global reflection of RV function but can be hard to measure accurately due to difficulty in identifying the endocardium in the heavily trabeculated RV. Pulmonary vascular resistance (PVR) may be estimated non-invasively by Doppler echocardiography using the ratio of the peak tricuspid regurgitant jet velocity (TRV) to the time velocity integral of the right ventricular outflow tract; PVR estimated by this method correlates relatively well with invasive measurements.[5] Using this technique, this patient's estimated PVR at the time of hemodynamic compromise was >5 WU and decreased by ~50% after thrombolytics. A unique RV contractile pattern has been described during acute PE whereby the free wall is hypokinetic but RV apical contraction is spared (“McConnell's Sign”).[6] However, this finding is insufficiently sensitive for acute PE and does not differentiate acute PE from RV infarction.[7] Other observations such as a pulmonary acceleration time below 60 m sec and PASP less than 60 mmHg (“60/60” sign) and mid-systolic notching of the flow velocity curve in the right ventricular outflow tract may be specific for elevated RV afterload (as in acute PE) but lack adequate sensitivity.[8] This case demonstrates the utility of adding quantitative measures to the echocardiographic assessment of PE. Quantitative measures remove inter-observer variability and allow serial measurements to be compared over time to assess for improvement or decline in function, as was the case for this patient.
The patient's initial echocardiogram (two weeks prior to the suspected PE) showed normal right ventricular size and function. At the time of clinical deterioration, repeat imaging showed significant dilation and hypokinesis of the RV. These findings demonstrate two unique aspects of RV/pulmonary circuit physiology: (1) RV function is exquisitely sensitive to changes in afterload (as in acute PE); and (2) after an acute increase in afterload the unprepared RV can only generate a modest increase in systolic pressure. RV systolic pressure after acute PE will typically not exceed 60 mmHg, in contrast to the compensated and hypertrophied RV in chronic thromboembolic pulmonary hypertension in which peak systolic pressure may become near-systemic. The clinical suspicion for PE was supported, not only by the patient's rapid hemodynamic improvement after administration of t-PA, but also by the objective increase in RV systolic function as measured by TAPSE and S'. This improvement in RV contractility after treatment with t-PA is likely due to both improved coronary perfusion to the RV and a decrease in afterload as pulmonary resistance decreases.[9]