
Abstract
Endothelin-1 is a potent vasoconstrictor and a therapeutic target in pulmonary arterial hypertension. Endothelial cells are the physiological source of endothelin-1 but in vitro data from our group shows that interferons (IFNα, IFNβ or IFNγ) induce endothelin-1 in pulmonary vascular smooth muscle cells. IFNs are integral to innate immunity and their antiviral and immunomodulatory capability has been harnessed therapeutically; for example, IFNα plays a critical role in the treatment of chronic hepatitis C infection. However, in some patients, IFN causes pneumonitis and possibly irreversible pulmonary arterial hypertension. In this study, we found that of 16 patients undergoing a six-month course of IFNα therapy, two demonstrated considerably increased serum levels of endothelin-1. We propose that IFN therapy results in elevated levels of endothelin-1 in some patients and when clinically significant levels are reached, pulmonary side effects could ensue. This hypothesis can be easily tested in IFN-treated patients by measuring serum endothelin-1 levels and cardiopulmonary physiological parameters.
Endothelin-1 is a potent vasoconstrictor that is a critical therapeutic target in pulmonary arterial hypertension (PAH)[1] and is implicated in the development of pneumonitis.[2] Endothelin-1 is principally released from endothelial cells. However, in early work from our group, we showed that in addition to the endothelium, the underlying vascular smooth muscle of systemic vessels can be induced to release endothelin-1 when stimulated in vitro with the combination of TNFα and type II interferon, IFNγ.[3] We found that the same phenomenon occurs in vascular smooth muscle cells from pulmonary arteries.[4] Recently, we have shown that endothelin-1 release is also induced when human pulmonary artery smooth muscle cells are stimulated with type I IFNα or IFNβ.[5] IFNs have powerful actions within the immune system where they play key anti-viral, anti-bacterial, and anti-tumoral roles; as such, IFNs are viable therapeutic agents in a number of disease states.[6] By far the most common therapeutic use of IFN (subcutaneously administered IFNα) is as a vital component in the treatment of chronic hepatitis C virus (HCV) which affects approximately 170 million people globally. IFNα is also an important treatment for a range of both solid and hematological malignancies. IFNβ is used to treat multiple sclerosis, IFNγ has been shown to be efficacious in the treatment of chronic granulomatous disease, and IFNΛ is currently being developed as a potential new treatment for HCV.
IFN therapy is most commonly associated with largely tolerable side effects such as influenza-like symptoms and mild depression. While these tend not to limit treatment, some patients experience more serious pulmonary toxicity. Pneumonitis is a well recognized complication of IFN treatment that affects up to 0.3% of patients.[7] Indeed, there is clinical evidence accumulating of a link between IFN therapy and rare but serious, and sometimes irreversible, cases of PAH.[8,9] If our in vitro findings are translatable to patients exposed to IFN drugs, we might hypothesize that circulating endothelin-1 levels could increase. If clinically significant levels were reached, debilitating pulmonary vascular side effects could become apparent. Of relevance, in a clinical trial examining the pulmonary effects of IFN treatment, nearly half of the 391 patients exposed to pegylated IFNα2a therapy experienced a decline in lung function (diffusion capacity) of greater than 15%; for 57 patients this deficit was still present six months after completion of IFN therapy.[10]
Our hypothesis is as follows: Endothelin-1 is induced in vascular cells of a subset of patients receiving IFN therapy leading to pulmonary side effects. Our in vitro data suggest that IFN-induced endothelin-1 will be synergistically increased in individuals where TNFα levels are also elevated. While documented cases of PAH associated with IFN therapy are rare, there is a theoretical risk that some cases may emerge years after treatment, as occurred in patients taking appetite suppressants such as aminorex in the 1960s-1980s.[11] We suggest that if our hypothesis is correct, the consequences of IFN therapy on the pulmonary vasculature in the longer term need be considered. Endothelin-1 is relatively simple to measure in serum but it is worth noting that as the lungs clear endothelin-1, interpretation of serum levels can be difficult, particularly in respiratory disease. Notwithstanding this, elevated serum levels of endothelin-1 have been measured in a number of respiratory conditions including pneumonitis[12] and PAH[13] and have been correlated with disease severity. Thus, we suggest that IFN therapy will induce endothelin-1, resulting in increased serum levels and hence pulmonary toxicity, in susceptible patients who may well be those individuals with elevated TNFα levels.
By way of a proof of concept study, we analyzed endothelin-1 levels by ELISA (R and D Systems, Abingdon, UK) in serum samples of a group of 16 patients taking a course of IFNα2a treatment for HCV infection. We measured serum levels before IFN treatment and then while on therapy at designated time points over a six month period (Fig. 1, Table 1). HCV viral load was also measured at these intervals. Inclusion criteria included previously untreated genotype 1 HCV infection, age between 18 and 65, absence of other viral infection, and no evidence of cirrhosis on a recent biopsy. Respiratory symptoms were not monitored in these archival samples but no patient developed gross lung disease requiring clinical intervention. Interestingly, we found that in serum in two of the 16 patients studied, endothelin-1 levels showed a clear time-dependent increase in response to IFN therapy. In both of these cases, IFN therapy resulted in complete viral suppression by Week 8, which was maintained throughout treatment suggesting that the mechanism of endothelin-1 induction was related to IFN treatment and not due to the virus itself. Neither of these patients had any other comorbidities to which raised levels of endothelin-1 could be ascribed. Furthermore, in those patients where IFN therapy was unsuccessful and where viral loads were high throughout the duration of treatment, endothelin-1 levels were not raised.
Serum endothelin-1 (ET-1) levels (pg/ml) for patients 1-16
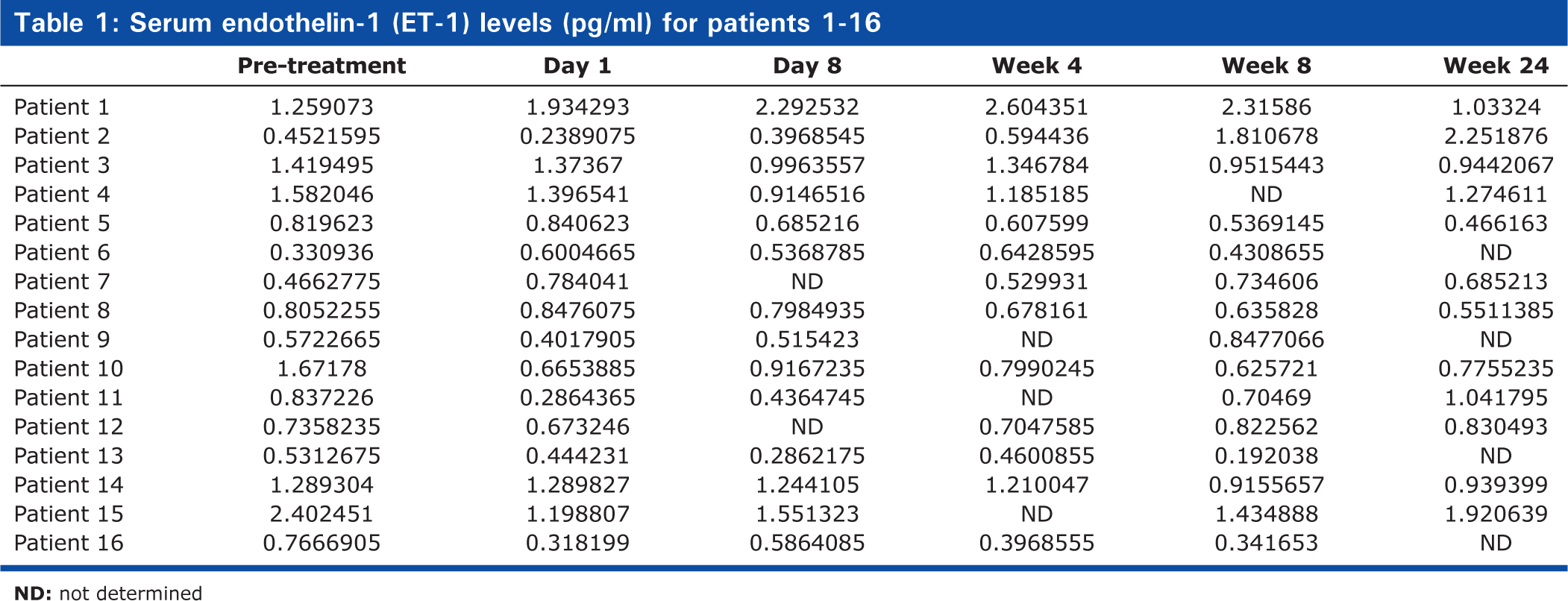
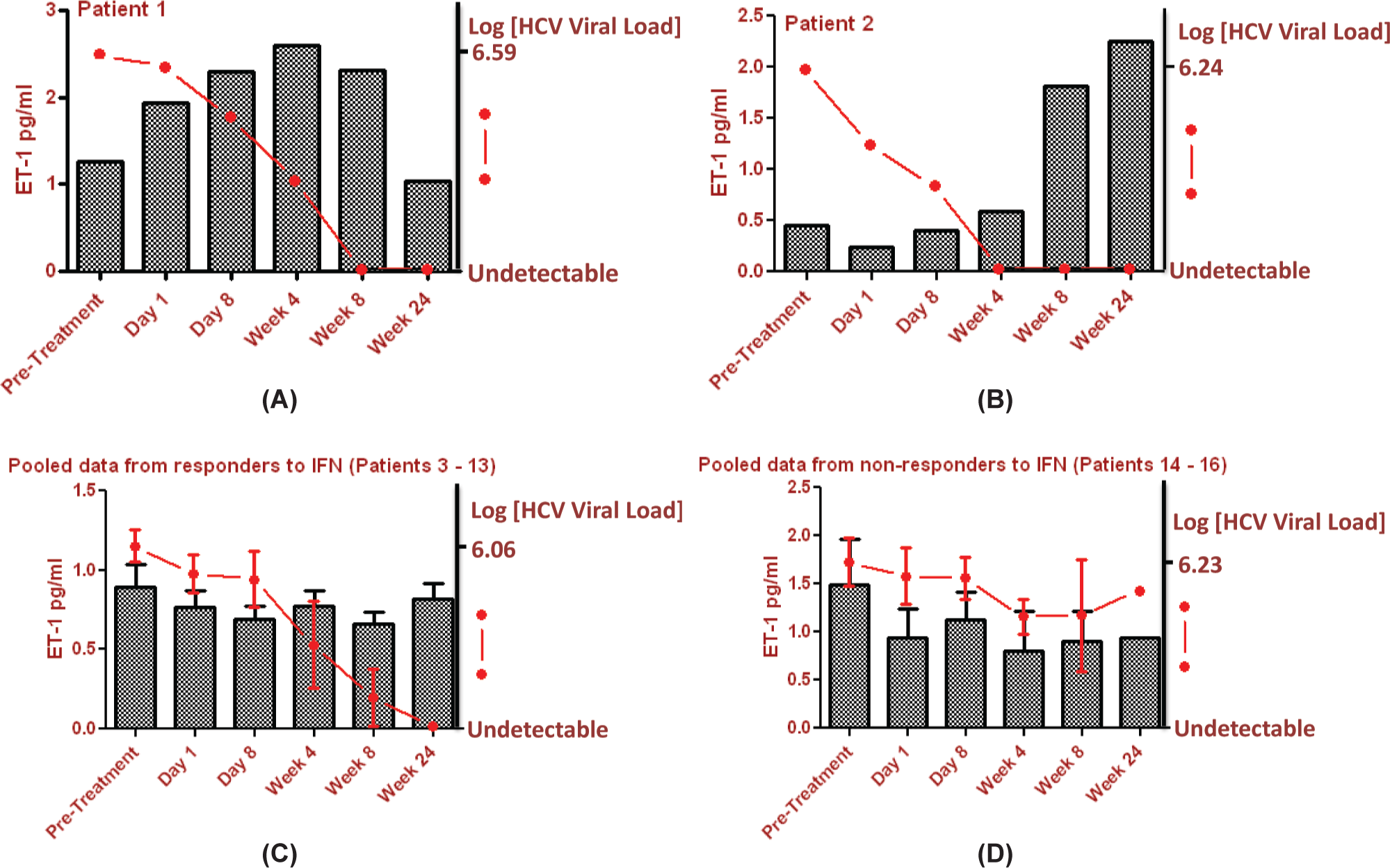
Serum levels of Endothelin-1 (ET-1) and hepatitis C viral (HCV) load in 16 patients receiving IFNα2a (180μg subcutaneously once weekly) and weight-based oral Ribavirin (1000-1200 mg daily) over a 6 month period. n = 16, M:F10:6, age range (mean): 24 years 5 months - 61 years 1 month (45 years, 2 months).
This is the first published report of an association between IFN and endothelin-1 in humans. As endothelin-1 is such a potent endogenous vasoconstrictor, this finding is significant and may represent a plausible mechanism for IFN-induced pulmonary toxicity. The symptoms of pneumonitis and in particular PAH develop insidiously and, if not specifically inquired about, often remain undiagnosed until cardiopulmonary physiological parameters are severely deranged. This is likely to progress slowly over a period of many months and so it is therefore unsurprising that none of these patients were diagnosed with fulminant respiratory disease requiring intervention during the study period. It is important to note that in many of the reports of IFN related PAH, diagnoses were made after therapy had been completed and were at that point irreversible.[9] Our finding that only a minority (two of 16) of patients sampled were affected is in keeping with the clinical picture of IFN-induced pulmonary toxicity where only a proportion of exposed patients develop clinical sequelae.
There exists much debate regarding the underlying pathological mechanisms linking certain viruses such as HIV with PAH. Our findings may help to explain this by providing a link between the innate immune response to chronic viral infection and production of host derived IFN with pulmonary vascular disease, as opposed to implicating viruses per se. As compared to the general population, patients infected with HIV are 2,500 times more likely to develop PAH and incidence is conservatively estimated at 0.5%.[14] While some investigators implicate viral proteins as being noxious to the endothelium, the virus itself has never been isolated in the pulmonary vasculature of affected individuals. There is chronic production of IFNα in patients with HIV infection[15] and increased IFNα expression correlates with HIV disease progression.[16] To our knowledge, no studies to date have attempted to look for a correlation between IFNα levels and PAH in HIV but a recent study of 116 HIV-infected individuals found raised levels of IFNγ, which correlated with increasing pulmonary artery systolic pressures.[17] We propose that this phenomenon may be unique to chronic viruses, which do not induce tolerance (such as HIV) and where there is continued activation of the host innate immune system. Furthermore, in systemic sclerosis, an autoimmune condition where the prevalence of PAH is estimated to be high as 12%[18] and where pathological pulmonary abnormalities have been found in over 50% of patients at autopsy[19] there is continued type I IFN activation and a positive correlation has been found between IFNα and pulmonary inflammation.[20]
We therefore suggest that IFN, whether produced endogenously in response to viral infection or autoimmunity or whether administered therapeutically, can target the pulmonary vasculature of certain individuals causing toxicity that might result in pneumonitis or even irreversible PAH and that this is driven in part by endothelin-1. We suggest that endothelin-1 levels should be measured and correlated with pulmonary function testing and echocardiography in patients receiving IFN therapy. Our aim is to determine whether sustained increases in endothelin-1 levels in serum correlate with a decline in lung function, rising pulmonary arterial pressures, and/or symptoms. If an association was found, endothelin-1 would become an important biomarker in the identification of pulmonary side effects related to IFN therapy. Endothelin-1 receptor antagonists are used routinely for the treatment of PAH and it is therefore tempting to speculate that these drugs may be clinically useful as prophylactic or rescue therapies for some patients receiving IFN therapies. In addition, if it is indeed those patients with higher circulating levels of TNFα that are at risk of developing IFN-induced pulmonary toxicity, one could again postulate that treatment with anti-TNFα agents could be beneficial. This work has potential far reaching clinical relevance to the millions of patients worldwide who have been exposed to or continue to receive IFN drugs. Furthermore, if our hypothesis is proven, we may have uncovered a novel clinical paradigm for the interaction between viruses, the innate immune system, and human disease.