
Abstract
Increased pulmonary blood flow (PBF) is widely thought to provoke pulmonary vascular obstructive disease (PVO), but the impact of wall shear stress in the lung is actually poorly defined. We examined information from patients having cardiac lesions which impact the pulmonary circulation in distinct ways, as well as experimental studies, asking how altered hemodynamics impact the risk of developing PVO. Our results are as follows: (1) with atrial septal defect (ASD; increased PBF but low PAP), shear stress may be increased but there is little tendency to develop PVO; (2) with normal PBF but increased pulmonary vascular resistance (PVR; mitral valve disease) shear stress may also be increased but risk of PVO still low; (3) with high PVR and PBF (e.g., large ventricular septal defect), wall shear stress is markedly increased and the likelihood of developing PVO is much higher than with high PBF or PAP only; and (4) with ASD, experimental and clinical observations suggest that increased PBF plus another stimulus (e.g., endothelial inflammation) may be required for PVO. We conclude that modestly increased wall shear stress (e.g., ASD) infrequently provokes PVO, and likely requires other factors to be harmful. Likewise, increased PAP seldom causes PVO. Markedly increased wall shear stress may greatly increase the likelihood of PVO, but we cannot discriminate its effect from the combined effects of increased PAP and PBF. Finally, the age of onset of increased PAP may critically impact the risk of PVO. Some implications of these observations for future investigations are discussed.
While increased pulmonary blood flow (PBF) has long been thought to provoke unfavorable pulmonary vascular remodeling,[1–5] the impact of wall shear stress in the lung is actually poorly defined, and most evidence indicates that increased PBF, in isolation, at most, weakly provokes pulmonary vascular obstructive disease (PVO) and might even be salutary. Evidence for this, starting with the indirect, includes the following:
If increased PBF causes PVO, it is presumably via increased wall shear stress, but investigations of the much better-characterized systemic circulation emphasize the pernicious effect of decreased and disordered shear, not when it is increased.[6–8] Indeed, experimental models of increased shear generally demonstrate an adaptive effect (e.g., increased luminal diameter) in systemic arteries.[7,8] Exercise, likely via increasing endothelial shear stress, increases NO signaling, reduces endothelin-1, and has multiple other anti-atherogenic effects.[8–10] Admittedly, most of this information pertains to large rather than resistance arteries, and to atherogenesis rather than intimal hyperplasia (but see Korshunov[11]), but if increased flow/shear acts to maintain a healthy phenotype in systemic blood vessels, why should this be different in the lung?
It is conceivable that exceedingly high shear could physically damage the endothelium, but it is unknown if this actually occurs with clinically relevant increases in blood flow.[12] Ghorishi and colleagues[13] used plastic casts of lamb lungs, normal and after creation of an aortopulmonary shunt, to model flow and shear stress. In shunted lungs with flow increased 3-fold, they found that shear stress in pulmonary arteries (PAs) < ∼250 μm diameter was high enough to possibly damage endothelium, but they found the same in normal lungs with normal flow for PAs < ∼70 μm diameter, making it difficult to know what to make of these data. Relevant direct observations are few, but a scanning electron microscopic study of five patients with increased PBF (but normal pulmonary arterial pressure [PAP]) due to congenital shunting lesions found the small PA endothelium was essentially normal.[14]
Even if increased shear stress does not damage the pulmonary vascular endothelium, could high PBF set up disordered shear in small PAs? The geography of small PA occlusion by intimal proliferation (often at branch points, or at the origin of lateral branches of small Pas[15–17]) might suggest disordered shear as an etiologic factor since atherosclerotic lesions occur in large systemic arteries at branching points;[8] however, intimal occlusion also occurs unrelated to branching points.[15,16] We know of no studies modeling the effect of flow on the pattern of shear in pulmonary resistance vessels, and it is unclear whether it is even plausible that resistance vessels in the lung could suffer oscillatory shear.
More directly and cogently, the vast majority of people with substantially increased PBF (two to four times normal) due to an atrial septal defect (ASD) have normal or only modestly increased PAP, even well into adulthood (Fig. 1; see “ASD: Increased PBF”). This alone strongly suggests that increased PBF does not reliably or powerfully actuate clinically significant pathological remodeling.
Mean PAP vs. the pulmonary to systemic flow ratio (Qp:Qs) in normal adults[54] and people with an ASD. With relatively few exceptions, PAP remains normal or only mildly elevated despite markedly increased PBF. Siltanen[156] (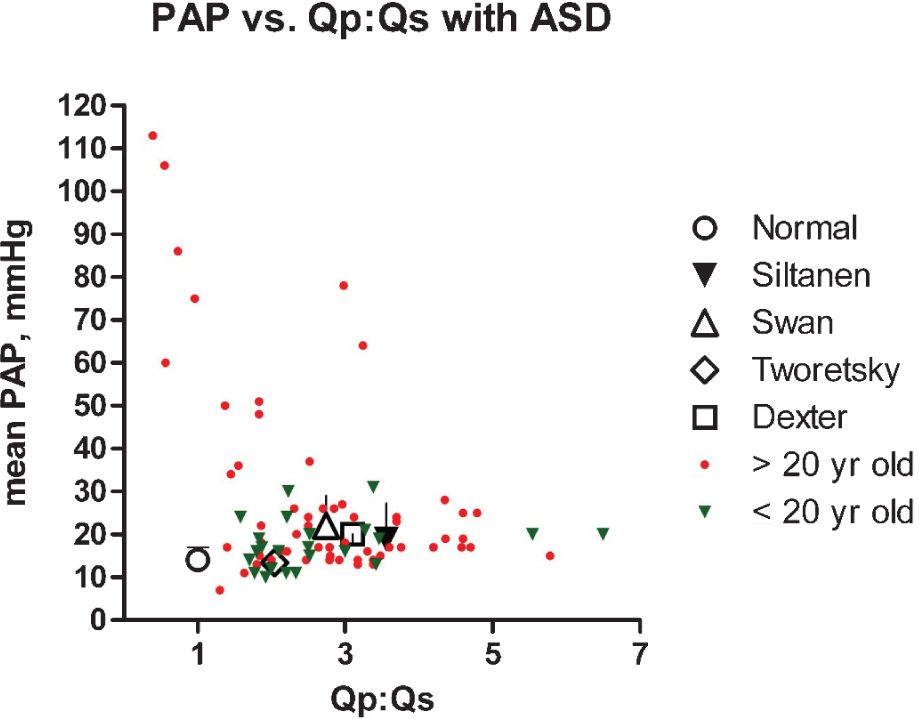
Other observations suggest that the lung is generally more flowophilic than flowophobic; the pulmonary circulation readily accommodates increased flow with exercise, usually showing a decrease in pulmonary vascular resistance (PVR) with increased flow.[18] Endurance athletes and even ordinary outdoor adventurers may spend several hours a day with more than twice normal PBF without obvious consequence. Indeed, a variety of in vitro and in vivo experimental models have shown that increased shear stress/PBF increases pulmonary NO signaling,[19–24] and possibly prostacyclin,[25,26] which presumably act to decrease PVR and possibly inhibit pathological remodeling. Indirect evidence for augmented endothelial NO release with chronically increased PBF in humans was provided by Celermajer and colleagues[27] who found that the ratio of acetylcholine to nitroprusside-induced pulmonary vasodilation is lower than normal in patients with increased PBF. The authors interpreted their data as showing endothelial dysfunction with increased PBF, but it is equally plausible that increased (but submaximal) baseline release of NO accounted for their findings. Taken together, these observations suggest that the lung” s talent for gracefully accommodating increased flow may actually make it resistant to damage from the same.
One might think that precisely defining the effect of increased shear stress in the lung would be an easy assignment for experimental models, but not so. Probably the best-defined preparation, exquisitely characterized over several years, has yielded much information; however, because both PAP and PBF are increased, the roles of shear and wall stress are inextricable[28] Furthermore, as with most other similar models, observations are made over a few months, not years, and intimal lesions—critical elements of PVO—are not observed. Most experimental models where increased PBF is the sole abnormality are in fact consistent with a flowophilic pulmonary circulation, but the periods of observation were much too short to determine the long-term consequences of increased shear stress (see “Animal Models of Increased PBF ± Increased PAP”).
But the arguments offered above are not definitive, and any theory of the effect of physical forces on the pulmonary circulation must account for the fact that increased PBF is, indeed, sometimes associated with PVO. Is there additional information that might be illuminating?
Observations of patients with cardiac malformations causing altered pulmonary hemodynamics yield insights, otherwise unavailable, regarding the role that mechanical forces (shear stress and circumferential wall stress) play in stimulating PVO. These clinical “models” reveal the effect of increased PAP and PBF over years to decades, and include lesions causing isolated alterations in pressure and flow, thus affording clues as to how these variables act independently. We therefore examined information (natural history, hemodynamic, and histological) derived from people having lesions which impact the lung in three distinct ways: (1) atrial septal defect (ASD; increased PBF with initially normal PAP); (2) mitral valve disease causing left atrial hypertension (increased PAP with ∼ normal PBF); and (3) large ventricular septal defect (VSD; increased PAP and PBF). Analyzing this information and published experimental studies, we asked the following: (1) Does increased PBF (i.e., absent increased PAP) stimulate PVO?, and (2) Do other factors, e.g., increased PAP, alter the effect of increased PBF relative to pathological remodeling?
HOW PVO IS DEFINED IN THIS PAPER
We focus on PVO caused by intracardiac shunting lesions, and use a functional (rather than histological) definition: a person with PVO has an increase in PVR sufficient to preclude safe closure of the intracardiac shunt, or PVR does not decline (indeed, it often progresses) if the defect is repaired (PVR = mean PAP − mean LAP/PBF, where LAP = left atrial pressure). This definition is not as sharp as one would like (clear criteria for operability are lacking, and there is no unequivocal level of unacceptably high PAP after defect closure), but it was chosen for the following reasons: (1) we are concerned here with the major clinical manifestation and liability flowing from pathologically remodeled vessels—severely and persistently or progressively increasing pulmonary hypertension (PH), not vascular changes with subtle or no clinical consequence (e.g., mild medial hypertrophy); (2) there are enough hemodynamic and natural history data for patients with cardiac defects to derive useful generalizations regarding the correlation of increased PAP/PBF and PVO as defined here. While there are a few reports correlating hemodynamic and outcome data with histological changes,[29–32] this information is too sparse to use pathological findings as an endpoint for this sort of analysis; and (3) most importantly, pathologic changes (i.e. medial hypertrophy, intimal hyperplasia, and even more advanced lesions) correlate only imperfectly with PAP, much less with fixed or progressive pathological remodeling.[29,31–35] It is not clear what a meaningful histological definition of PVO would be given that the mildest abnormalities (distal extension of smooth muscle) are usually of little consequence, and the iconic lesions of irreversible PH (plexiform) are not invariably present with severe disease.[32,34]
But note that the lack of PVO does not necessarily imply an absence of pathological remodeling, only that the circulation is not so altered as to suffer permanent or progressively increased PVR. Also intimal hyperplasia/fibrosis is taken as being an essential feature of PVO (medial hypertrophy being reversible[33]), which is important when evaluating animal models.
ASD: INCREASED PBF
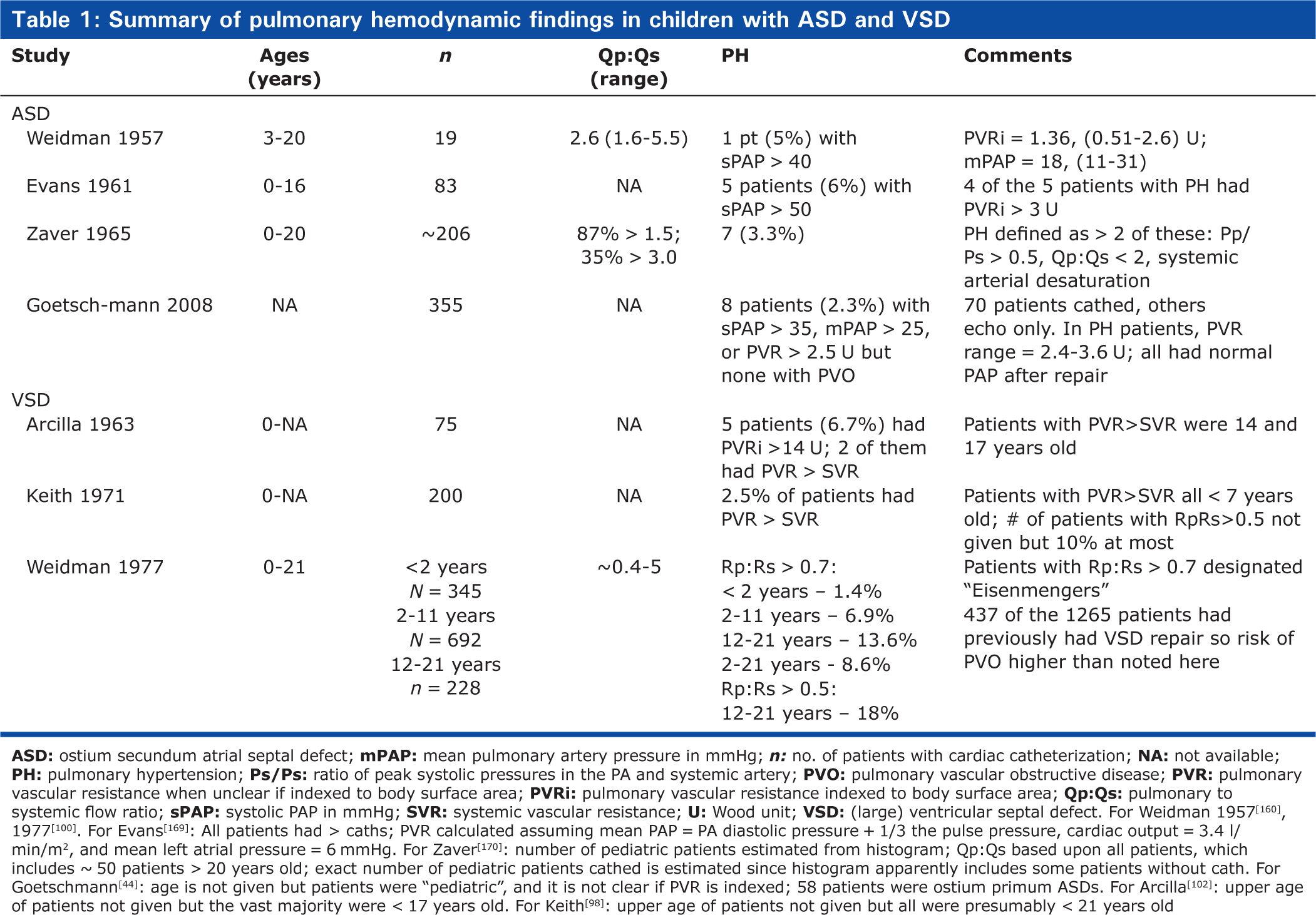
People with a sizable ASD develop a large left-to-right shunt and occasionally develop PVO, but unlike patients with left atrial hypertension or a large VSD, they seldom have significantly increased PAP, much less PVO, until the third decade of life. The risk of increased PVR (>3 Wood units) with ASD in children (before age 20 years) is < ∼3% (Fig. 1 and Table 1).[36–40] Indeed, PH in children with an ASD is so uncommon as to occasion reports of such, and it is unclear whether the presence of the ASD was pathogenic in causing PH in these often very young patients.[39,41] The risk of PVO is surely much lower in children; even in adults, most patients with modest, and some with severe, PH show a marked decline in PAP after repair of the ASD (Fig. 2).[42–44] Estimates of the lifetime risk of developing significant PH vary greatly, but ∼10–15% seems reasonable[38,45,46] although, again, this overestimates the risk of PVO. There is a single report of a large number of children with ASD and increased PAP and PVR.[47] About 40% of 423 patients <20 years old had systolic PAP > 32 mmHg, ∼11% having total PVR > 5 units (i.e., LAP was not subtracted from PAP), and 6.8% of patients <10 years old had PH with right-to-left shunting. No other hemodynamics were provided, nor whether PVR was indexed. The authors acknowledged that these findings are atypical, and speculated that they may be related to recurrent respiratory tract infections or ethnic variation in susceptibility to PH. It is difficult to know what to make of this report, except that it is inconsistent with other published data.
Summary of pulmonary hemodynamic findings in children with ASD and VSD
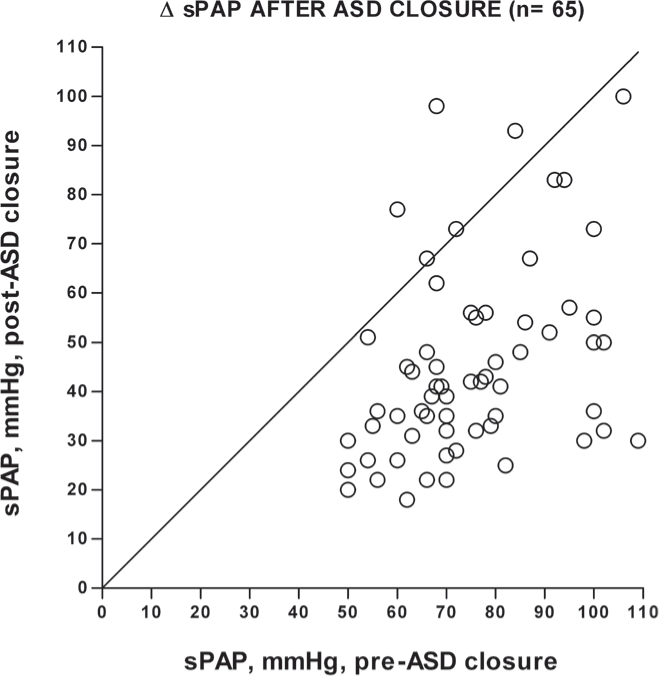
Systolic PAP (sPAP) before and after surgical closure of ASD in 65 patients with PH. Most patients had considerable reduction in PAP after repair, indicating that a large component of the pre-operative PH was not due to PVO. (Note that this figure overestimates the fall in PVR with ASD closure since most patients had increased PBF prior to operation.) Systolic PAP was used as mean PAP and PVR was not available in all studies. Data are from seven studies and include only patients with systolic PAP > 50 mmHg. Patients were all adults except in Cohn[163] and Walker,[164] which included an indeterminate number of children. Time from surgery to the post-operative cath was 2 months to 10 years, except in one patient (2 weeks). Data do not reflect intra- and postoperative deaths, and underestimates the incidence of progressive PH since in some patients PAP increases with time after ASD closure.[158,158,163,165–168]
Histological findings confirm that, in most patients, decades of increased PBF has little obvious impact on small PAs; patients with ASD and normal PAP, even those with large shunts, have essentially normal small PAs,[48,49] although morphometric analysis has revealed distal extension of smooth muscle in children with increased PBF and normal PAP.[50] Dilation of small PAs in patients with moderate PH was noted by Edwards.[48] High-grade lesions (> Heath Edwards grade IV) occur in the small minority who have increased PVR.[39,41,48,51]
Two reports suggest that the incidence of PH with ASD may be higher in children residing at increased altitude (1340–1600 m).[52,53] Neither study is definitive, and it is perhaps surprising that this moderate increase in elevation—which does not appreciably increase PAP in normal adults[54]—should have much impact, but these reports resonate with an interesting experimental study (see “Animal Models of Increased PBF ± Increased PAP” and “Limitations and Possible Implications”).
Total anomalous pulmonary venous connection
Total anomalous pulmonary venous connection (TAPVC) without pulmonary venous obstruction is similar to an ASD insofar as PBF is increased due to an atrial-level shunt; however, this defect is frequently attended by markedly increased PAP, and even intimal hyperplasia in the first few months of life.[55–58] It appears that PH in such patients is often present in the first few postnatal days; of the 13 consecutive neonates with unobstructed TAPVC at our institution, all but one had estimated RVP (by echocardiography) > one-half systemic, and six were systemic or suprasystemic, between 3 and 14 days of age. These data are consistent with the following interpretation: unlike an ASD, with unobstructed TAPVC, substantially increased PBF must pertain at birth, since cardiac output is limited to whatever systemic and pulmonary venous blood returned to the right atrium finds its way across the (often somewhat restrictive[58]) atrial septal opening. Because the newborn pulmonary circulation has a limited capacity to dilate with increased flow (as reflected by its anatomy,[59] and the fact that PAP is relatively high for at least the first 24 hours[60]) it cannot accommodate markedly increased PBF at low pressure. PH is therefore present from birth and tends to remain so (see “Limitations and Possible Implications”). At any rate, given how early PH occurs with TAPVC, it seems exceedingly unlikely that this is due to flow-incited pathological remodeling.
MITRAL VALVE DISEASE: PH DUE TO PULMONARY VENOUS HYPERTENSION
PH due to mitral valve disease affords a unique window on the impact of increased PAP on blood vessel structure and function. This variety of PH is caused by high LAP, not external stimuli (e.g., alveolar hypoxia or toxins), pulmonary parenchymal abnormality, or systemic ailment (e.g., collagen vascular disease). Because cardiac output is normal or reduced,[61–65] increased PBF is not a factor. And while the impact of pulmonary venous hypertension on PVR is variable, it is very unlikely that the increased PVR reflects inherent abnormality of pulmonary vessels (pace idiopathic PH) since it is so commonly seen with mitral valve disease, and reduction of LAP usually causes reduction or resolution of the PH. Indeed, this is one of the few forms of PH for which the stimulus can be cleanly eliminated, and many hemodynamic studies of patients with mitral valve disease—conducted before and after the relief of left atrial hypertension—have yielded much information regarding its characteristics.
PAP is increased with left atrial hypertension, in some cases only due to passive transmission of LAP (hence calculated PVR is unchanged), but in others PVR is elevated. The increase in PAP and PVR is highly variable, but often considerable. In five reports of adults undergoing intervention for mitral valve disease (
The increased PVR is due to pathological remodeling and active vasoconstriction. The pathological remodeling consists of adventitial thickening, medial hypertrophy, distal extension of smooth muscle, longitudinal muscle bundles in the media, and intimal thickening and fibrosis.[33,72–75] Higher grade lesions (e.g., dilatation and plexiform) can be part of remodeling but are uncommonly seen.[72,76] Vasoconstriction is revealed by an acute fall in PAP/PVR with vasodilators (intravenous acetylcholine, hexamethonium, priscolene, and inhaled NO). The response to these agents is variable, and not all patients show vasodilation, but a decrease of ∼40% in PAP/PVR is typical.[67,77–79] Vasoconstriction is also inferred from the fact that PVR falls within hours of mitral valve intervention, followed by a further decline over months, the latter presumably reflecting remodeling of pulmonary vessels.[64–66,80] In four series of balloon mitral valvuloplasty (totaling 114 patients), the percentage fell in PVR immediately after the procedure was 24+8%, and several months later the PVR was 51+9% lower than baseline.[64–66,81]
Indeed, despite having PH for many years, adults and older children[73,75,76,82] with mitral valve disease who have reduction of left atrial pressure with valve repair reliably (albeit not invariably) show a substantial fall or even normalization of PVR (Fig. 3); even patients with mean PAP > 50 mmHg generally show a large reduction in PAP and PVR (Fig. 3).[63,81,83,84] Almost all hemodynamic data are for < ∼1-year follow-up, but one large series found that the reduction in PVR lasted several years.[85]
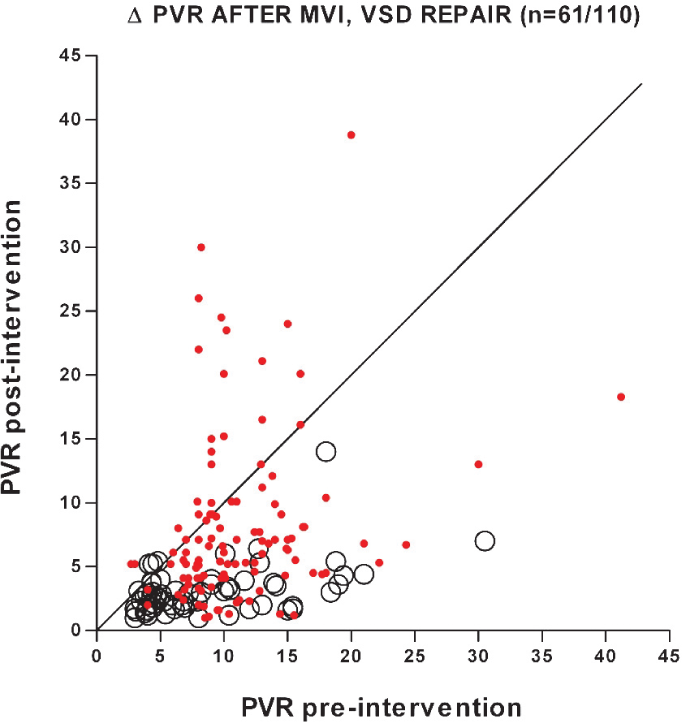
Change in PVR after mitral valve intervention (MVI) to effect relief of mitral stenosis/regurgitation (circles,
The reliable reduction of PVR with mitral valve replacement or repair is probably also true in the first few years of life,[75,76,86] although the available data are limited. We are aware of little data regarding the effect of relief of left atrial hypertension in patients with a longstanding increase in PVR due to left atrial hypertension present since birth. As discussed in “Limitations and Possible Implications,” this may be important.
As noted above, PVR likely declines partly due to remodeling. Smooth muscle hypertrophy/hyperplasia presumably regresses, interstitial edema in the vessel wall is perhaps reduced, and intimal hyperplasia and fibrosis may regress as well, although direct evidence for this is scant.[33] It is interesting that intimal lesions do not imply PVO in many mitral stenosis patients;[72,73,75] presumably they are less extensive or occlusive than those with high pressure/flow defects.
PH WITH HIGH ALTITUDE RESIDENCE
The idea that increased PAP alone, even if longstanding, seldom provokes PVO is confirmed by observations of people living at high altitude.[87–90] PH in high-altitude dwellers is usually modest (< ∼30 mmHg), but can be severe, especially with exercise;[91,92] yet, PAP normalizes with relocation to low altitude.[87,88,90] The reversible nature of high-altitude PH is presumably related to the fact that remodeling with alveolar hypoxia is limited to medial hypertrophy and distal extension of smooth muscle.[93–96]
VSD: INCREASED PAP AND PBF
With an unrestrictive VSD, systolic PAP = systolic aortic pressure since birth, and PBF is increased early on. Because PBF is largely a function of PVR with a large VSD, patients with severely elevated PVR have normal or even reduced PBF, but most patients who ultimately develop PVO had a pulmonary to systemic flow ratio (Qp:Qs) >2, prior to the evolution of PVO.[97–100] There may be something like a threshold PAP for the development of PVO with a VSD; two reports suggest that it seldom develops with mean PAP < 50 mmHg,[100, 101] at least in childhood.
The age-related risk of PVO with a large VSD is unknown since its natural history is often modified by surgical intervention or death, and relevant information from the pre-surgical era is sparse. It is clear that, as with an ASD, the risk of PVO increases with age, but with a VSD PVO is much more common in childhood. While the first 2 years of life pose little risk of PVO,[29,100,102–104] its incidence increases substantially thereafter. Reports vary regarding the prevalence of PVO, but the largest (1,265 patients) and perhaps most representative study (by virtue of being prospective, multi-center, and presumably reflective of all VSD patients seen at the participating centers) reported severe elevation of PVR (in many cases with reversal of shunt) in 8.6% of patients ages 2–21 years[100] (Table 1). This is surely a substantial underestimate of the risk of developing PVO as one-third of the patients had previously had repair of the defect, many with high PAP at the time of surgery. And with VSD, unlike left atrial hypertension, high PVR often reflects PVO, even in childhood. Figure 3 depicts pre- and post-operative PVR in patients <18 years old at VSD repair; for many, resistance remained high or increased after operation. This figure under-represents the true incidence and severity of PVO in operated patients since those who died at or after repair due to PVO are not accounted for, and some patients with modest elevation in PVR at first post-operative study doubtlessly had a further increase with time.[105–108]
Histological findings are consistent with the clinical picture. For patients < ∼2 years old with a large VSD, medial hypertrophy and distal extension of smooth muscle are nearly invariably seen and a reduction in the number of small arteries is often noted; intimal lesions are much less common and advanced lesions are rare. As age advances, so does the likelihood of intimal hyperplasia and fibrosis, plus pre-dilation, dilation, plexiform, and other high-grade lesions.[29,32,50,109–111]
The role of increased LAP with VSD
Patients with a large VSD may have increased LAP, which could possibly contribute to the tendency to develop PVO. If so, this contribution is likely small—in four reports of 43 children with a large VSD (Qp:Qs > 2 and/or congestive heart failure), mean and median left atrial pressure were 11.7 + 4.52 mmHg and 11 mmHg, respectively[97,112–114] which is only modestly higher than normal (8 + 2 mmHg60).
Too much of a good thing?
With virtually all models of substantially increased PBF, human and experimental, both PBF and PA O2 saturation are increased; might increased PA blood O2 saturation, then, actually be the factor that provokes PVO? Assuming an arteriovenous O2 saturation difference of 20% and pulmonary venous and systemic arterial O2 saturations = 95%, if Qp:Qs = 2, PA O2 saturation = 85% (pO2 ∼50 mmHg). Normal PA pO2 is ∼ 40 mmHg, and it is conceivable that this modest difference in PA pO2 could influence elaboration of substances involved in pathological remodeling. (Patients with d-transposition of the great arteries and VSD have PA O2 saturations yet higher (> 90%) and—consistent with the high PA O2 sat/remodeling hypothesis—a tendency to develop PVO much earlier than patients with VSDs.[115–117]) While there are rare situations where increased PBF co-exists with normal PA O2 saturation (e.g., isolation of the left or right PA, or surgical pneumonectomy), the data from existing models are too few to answer this question.
There are, however, cogent arguments against the notion that high PA O2 saturation causes PVO: (1) As noted above, alveolar hypoxemia (arterial pO2 < ∼ 60 mmHg)[118] stimulates PA distal extension and hypertrophy of smooth muscle;[93–95] why might exposing small PAs to modestly higher than normal pO2 stimulate the same? and (2) with high pressure shunting defects, early events in vascular remodeling (distal extension and hypertrophy of smooth muscle, and the initial appearance of intimal proliferation) occur in PAs < 150 μm in diameter.[15,34] Since O2 from alveoli (pO2 = 100 mmHg) diffuses through the vessel wall and into the blood of PAs this diameter,[119,120] it seems unlikely that PA blood pO2 of 50 or even 60 mmHg could much influence the biology of the cells in these small vessels.
ANIMAL MODELS OF INCREASED PBF + INCREASED PAP
Increased flow with little or no initial increase in PAP has been effected by left pneumonectomy, ligation of the left or right PA, creation of an arteriovenous fistula, or both. The effects of left pneumonectomy or left PA ligation (increasing flow to the right lung ∼75%) or an arteriovenous fistula are fairly consistent, although there is some variability in findings. PAP is mildly increased in most but not all animals, roughly in line with the increase in PBF, although some animals show a modest disproportionate increase in pressure after several months.[121–128] Medial hypertrophy is sometimes observed, while intimal lesions are not.[121–124,126,128] Dilation of small PAs was demonstrated in two studies, but only at isolated time points.[122,128] The absence of PVO or even severe PH could be related to the very short duration of these studies (< 18 months except for Davies,[126] which was 5 years) and the fact that with pneumonectomy or left PA ligation PBF is much less than with a large ASD.
But one provocative study, using left or right PA ligation in newborn calves (one to two days after birth), found that increased PBF was much more consequential.[129] With left PA ligation at sea level, there was at most mild PH even after four years, but at 1,600 m elevation severe PH developed in < 2 months. Right PA ligation in the first 24 hours of life (but not at one week of age) produced severe PH, even at sea level, in four of six animals. The fact that calves are more reactive to alveolar hypoxia than most species,[130] and that PA ligation was performed shortly post-natally (see “Limitations and Possible Implications”), may explain these findings, although it is interesting that these experiments resonate with the human observations of ASDs at elevation noted in “ASD: Increased PBF.” The early development of severe PH with the combination of increased PBF and alveolar hypoxia may be the key, and is discussed further in “Limitations and Possible Implications.”
Increased PBF and PAP has been produced in animals by placing a shunt between the aorta and the main or branch PA (usually using the subclavian artery or a prosthetic tube graft), or attaching a branch of the PA to the aorta. Moderately increased PAP (mean PAP ∼30–45 mmHg) and flow, produced by a restrictive aortopulmonary connection, cause medial hypertrophy and distal extension of smooth muscle in as little as one month;[131–134] whether intimal lesions would eventually develop is unknown, owing to the limited duration of these studies. With severely increased PAP (mean PAP >50 mmHg) and flow, intimal hyperplasia occurs in as little as two months, proceeding to obliteration of small PAs within a few more.[131,135–141] But whether these models accurately reflect the clinical biology of vascular remodeling is perhaps doubtful; in nearly all (except, perhaps, two studies[131,140]), the pulmonary circulation is subjected to an abrupt and marked increase in PAP and PBF at the time of shunt creation. This causes acute and severe vascular injury (edema, hemorrhage into the PA wall and into the lung, inflammatory cells in the sub-intima and adventitia, and necrosis).[137,138] Most of the models with very high PAP at shunt creation may therefore better reflect the response to acute injury than chronically increased pressure and flow.
In summary, animal models of increased PBF without transmission of systemic pressure into the PA indicate little tendency toward significantly increased PAP, even after five years, the Vogel paper excepted.[126,129] Medial hypertrophy is often observed, but not intimal hyperplasia, implying that increased PBF is not a powerful stimulus for PVO. However, the short duration of experiments (relative to the evolution of human PVO) and modest augmentation of PBF (in most studies) make this interpretation tenuous at best. Studies showing intimal proliferation with increased flow and very high PAP suggest the importance of pressure in PVO generation; however, most of these studies may be better models of acute injury than chronic shunting lesions.
SUMMARY AND ANALYSIS OF CLINICAL AND EXPERIMENTAL OBSERVATIONS
Cardiac shunting lesions with increased PBF but low PAP have only a small tendency to develop PVO (< 15%), which evolves very slowly. Isolated left atrial hypertension increases PAP and (less commonly) substantially increases PVR; relief of the LA hypertension usually, although not invariably, causes PVR to decline markedly, often to normal or nearly so. This is true even in older adult patients who presumably have had PH for many years. Patients with high PAP (> ∼ 50 mmHg) and increased PBF are at much higher risk for developing PVO in the first two decades of life than are patients with high PBF only. Experimental shunting models are generally consistent with these findings, although none recapitulate the clinical condition of longstanding, gradually developed, very high pressure and increased flow.
The effect of increased PBF and PAP on endothelial shear stress in each of these three clinical “models” can be further considered. Systemic arteries generally respond to chronic alterations in flow by remodeling to adjust their radius to return wall shear stress to normal.[142–144] It turns out that the lung's circulation is not efficient at normalizing shear with increased flow, especially with high pressure, as is apparent when considering an infant with a large VSD. As inferred in “VSD: Increased PAP and PBF,” the typical “pre-PVO” VSD patient in the first year or so of life will have a mean PAP of >50 mmHg and a Qp:Qs > 2. The calculated PVR is therefore high (Table 2). If PVR is proportional to the square of the cross-sectional area of the pulmonary vascular bed, the area must be decreased relative to normal, and the resistance vessels are therefore reduced in number, diameter, or both. Histological observations in patients with high pressure and flow cardiac defects are consistent with the following: although findings vary, with a VSD the number of small PAs is often reduced,[34,50,111,145] and the external diameter of small PAs normal or reduced much more often than dilated. Even when the external diameter is normal, the luminal diameter may be reduced by virtue of medial hypertrophy[34,50,111,145] (likewise, in multiple experimental models of increased PBF + PAP small PAs are usually of normal or reduced diameter[122,132,140]). Wall shear stress is presumably increased with elevated PVR but normal PBF,[146] but to a much greater degree when both PVR and PBF are increased. This is true whether all parallel resistance vessels are reduced in diameter or (much more likely) some vessels are of reduced diameter (or occluded) while others are unaffected.
PVR in the normal lung, with left-to-right shunting lesions, and with mitral stenosis
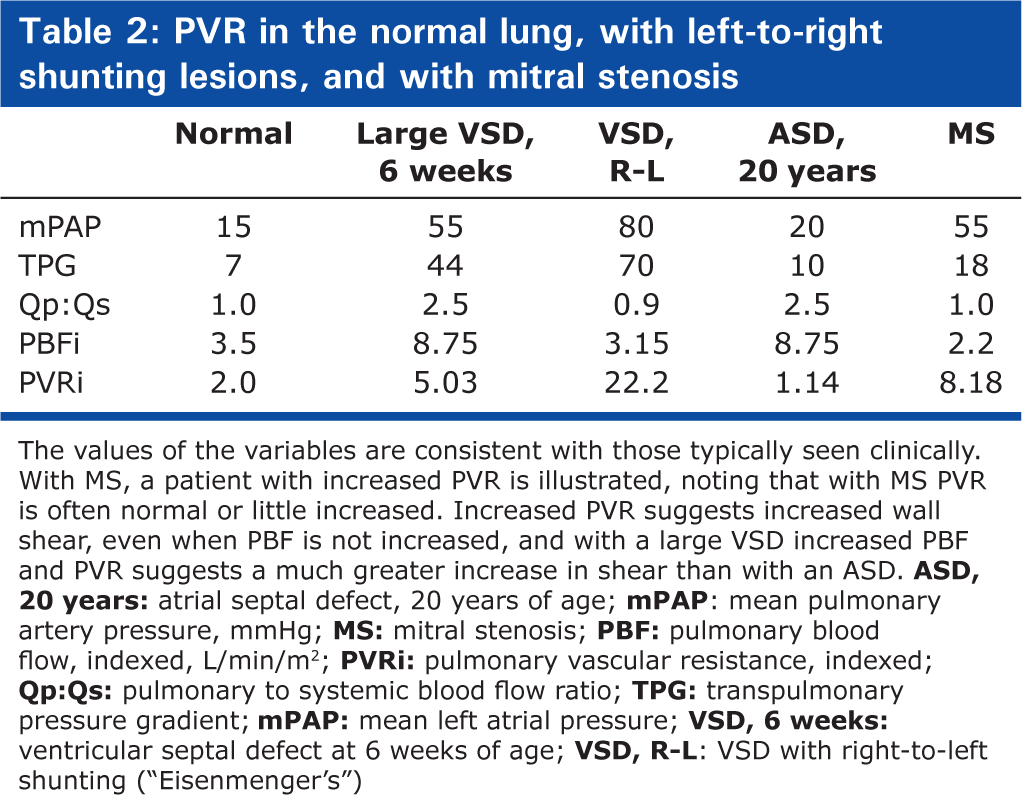
The values of the variables are consistent with those typically seen clinically. With MS, a patient with increased PVR is illustrated, noting that with MS PVR is often normal or little increased. Increased PVR suggests increased wall shear, even when PBF is not increased, and with a large VSD increased PBF and PVR suggests a much greater increase in shear than with an ASD.
LIMITATIONS AND POSSIBLE IMPLICATIONS
This analysis has multiple limitations: (1) As noted in “How PVO Is Defined in this Paper,” the phenomenon of interest (PVO) is defined functionally, and the edges are fuzzy rather than sharp; (2) The risk of developing PVO with the clinical models examined here is only imprecisely defined, (but the clinical manifestations are different enough to imply clear differences in the biology of increased flow, pressure, and their combination); (3) This analysis does not account for all phenomena related to cardiac lesions with altered pulmonary hemodynamics (e.g., the unusually rapid development of vascular disease with d-transposition of the great arteries with VSD); and (4) Finally, this analysis—which is mostly focused on hemodynamic rather than cellular and molecular events—might seem a bit jejune, given that current investigations have advanced beyond physiology and into elegant biochemical studies of shunted animals. But we believe that it is crucial to understand as best as possible the effect of pressure and flow alterations in the organism of most interest if interpretation of experimentally derived data is to be most useful. With these limitations in mind, we suggest the following:
Increased shear stress attends atrial level left-to-right shunts, and when PVR is increased due to left atrial hypertension, both of which are associated with low, slow risk of PVO, indicating that increased shear stress, as an isolated variable, at most weakly provokes significant pathological remodeling. In fact, the constellation of increased wall shear and PH but low risk of PVO with pulmonary venous hypertension is consistent with the possibility that increased shear actually reduces the tendency for PVO. While these observations only weakly infer this, investigators ought to consider the possibility that increased flow/shear may inhibit pathological remodeling when designing and interpreting experiments. The strong bias toward assuming that increased shear in the lung must be pernicious may reduce the chance that any favorable effect of increased shear will be uncovered or interpreted as such—you are most likely to find what you look for.
Greatly increased shear stress may be another matter. Increased PBF at high pressure and resistance implies much higher shear and much greater risk of PVO. (Indeed, in patients with increased PBF and PH, endothelial denudation[111] or alteration of endothelial surface characteristics and cytoplasmic components[41] has been observed). Clinical and experimental observations do not reveal, however, whether it is very high shear per se, or the combination of increased shear and high pressure which increases the risk of PVO. The notion that increased pressure and flow act synergistically is an attractive way to explain the high risk and rapid development of PVO with a large VSD.[49,75,147,148] But there is a third possible explanation for the apparent confluence of high pressure, flow, and risk of PVO, which is offered below (see #4).
Patients with an ASD do develop PVO often enough that it seems likely that increased PBF can predispose to or provoke pathological remodeling in some people or circumstances. What other factors might conspire with increased shear to cause PVO? The usual suspect, an undefined and uncommon “innate predisposition,” is likely relevant. As noted in “Mitral Valve Disease: PH Due to Pulmonary Venous Hypertension,” a sub-population of adults with left atrial hypertension has a marked increase in PVR, and humans also show remarkable variability in the pulmonary vascular response to alveolar hypoxia.[130] It could be that a general predisposition to pulmonary vasoconstriction/remodeling, regardless of the inciting stimulus, is required for shear stress to participate in pathological remodeling.
It is also possible that identifiable environmental stimuli, e.g., alveolar hypoxia (see “ASD: Increased PBF” and “Animal Models of Increased PBF ± Increased PAP”) may predispose to PVO with an ASD. Increasing PBF by pneumonectomy or aortocaval shunt in rats given monocrotaline causes intimal lesions (which are not observed without increased PBF), suggesting that an additional factor(s) is needed for increased PBF to be harmful.[5,149] Since monocrotaline causes endothelial injury,[150] perhaps such injury (due to a viral insult, inflammation, or other cause), combined with increased shear, predisposes to PVO in people.[146] At any rate, the small effect of modestly increased shear stress (as an isolated factor) in the vast majority of humans suggests that experimental observations of the effects of increased shear may be especially informative if coupled with investigation of other possibly clinically relevant perturbations (e.g., hypoxemia, virally induced inflammation).
Very high PAP caused by left atrial hypertension leads to pathological remodeling but uncommonly to PVO. It is conceivable that left atrial hypertension somehow protects against development of fixed remodeling (e.g., perhaps by passively distending resistance vessels and thus reducing shear somewhat). We favor the notion that increased pressure is a weak stimulus for PVO, and that it is increased pressure plus flow that really matters, but knowledge of no data which definitively establishes this.
As was suggested decades ago,[150] the age of onset of PH may be of signal importance. With an unrestrictive VSD, PAP is markedly increased from the get-go. The normal postnatal thinning of resistance vessels is inhibited, and other developmental changes are altered.[151–153] With lower-risk lesions (ASD, left atrial hypertension), the vascular bed remodels from a baseline of normal structure and function (e.g. rheumatic mitral stenosis in an adult) or at most subtle changes in architecture (e.g., ASD). It could be that increased PAP and PBF imposed on a substrate of thick-walled resistance vessels, functionally immature and with reduced luminal area, is much more likely to ultimately cause intimal proliferation than the same perturbation in mature vessels. Indeed, this is the only obvious hemodynamic explanation for early development of severe PH in patients with large left-to-right atrial level shunts at birth (see “Total anomalous pulmonary venous connection”). Long-term observations of patients with severe PH since birth due to isolated pulmonary venous hypertension would be illuminating, but such lesions are poorly tolerated and survival usually short without effective surgical relief.
To understand the distinct effects of different mechanical forces on PAs, clinical and experimental observations of shunting lesions need to clearly define the hemodynamic perturbation(s) involved and interpretation of results need to be taken into account (i.e., in models with both increased PAP and PBF, attribution of changes in the vessel wall to one or the other may not be warranted). Moreover, since stretch impacts release of growth factors from vascular smooth muscle,[154,155] increased wall stress may alter endothelial function via its impact on smooth muscle—i.e., the effect of mechanical forces on PAs may not be a one-way street from endothelium to smooth muscle; attributing altered endothelial function to increased wall shear stress in the setting of increased PAP, therefore, may not always be correct.