
Abstract
Background:
Following the example of L-valine prodrugs of antiviral nucleoside analogues, L-valine ester of cyclopropavir (valcyclopropavir) was synthesized.
Methods:
The known tetrahydropyranylcyclopropavir was transformed to N-(tert-butoxycarbonyl)-L-valine ester, which was deprotected to valcyclopropavir.
Results:
Stability of valcyclopropavir towards hydrolysis at pH 7.0 roughly corresponded to that of valganciclovir. Valcyclopropavir inhibited replication of human cytomegalovirus (HCMV, Towne and AD169 strains) to approximately the same extent as the parent drug cyclopropavir. Pharmacokinetic studies in mice established that the oral bioavailability of valcyclopropavir was 95%.
Conclusions:
The prodrug valcyclopropavir offers some improved therapeutic parameters over the parent compound cyclopropavir.
Introduction
Efficacy of antiviral nucleoside analogues is often hampered by their low solubility and bioavailability. To address this deficiency, several types of prodrugs have been synthesized [1]. Amino acid esters were found to be especially advantageous. Valine ester prodrugs of acyclovir (valacyclovir [Valtrex] (
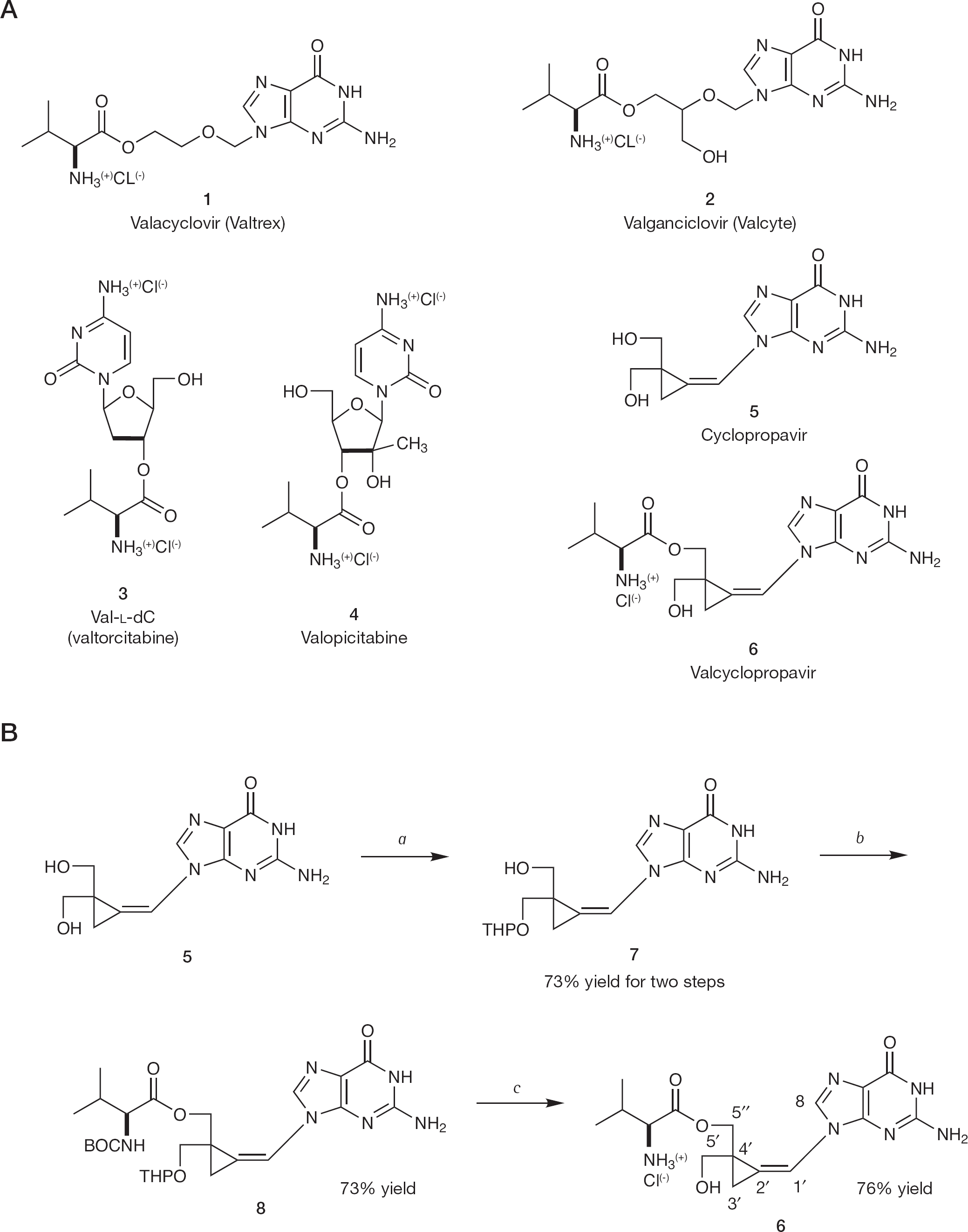
Valine ester prodrugs of some antiviral nucleoside analogues
In our laboratory, we have developed a new class of antiviral agents that are methylenecyclopropane analogues of nucleosides [13]. Among these analogues, cyclopropavir (
Methods
Synthesis
The mass spectra (MS) were determined in an electrospray ionization (ESI) mode. Valcyclopropavir (
(Z)-9-{[2-(N-tert-Butoxycarbonyl-L-valyloxymethyl)-2-(tetrahydropyranyloxymethyl)cyclopropylidene]-methyl}guanine (8)
A mixture of N-(tert-butoxycarbonyl)-L-valine (955 mg, 4.4 mmol) and dicyclohexylcarbodiimide (537 mg, 2.6 mmol) in dichloromethane (36 ml) was stirred at room temperature under nitrogen for 3 h. The insoluble portion was filtered off and solvent from the filtrate was evaporated in vacuo. The resultant N-(tert-butoxycarbonyl)-L-valine anhydride [(BOCVal)2O] was dissolved in N,N-dimethylformamide (DMF; 50 ml) and compound
(Z)-9-{[2-Hydroxymethyl-2-(L-valyloxymethyl) cyclopropylidene]methyl}guanine hydrochloride (6)
A solution of compound
Kinetics of hydrolysis of valcyclopropavir (6) at pH 7.0 A solution of compound
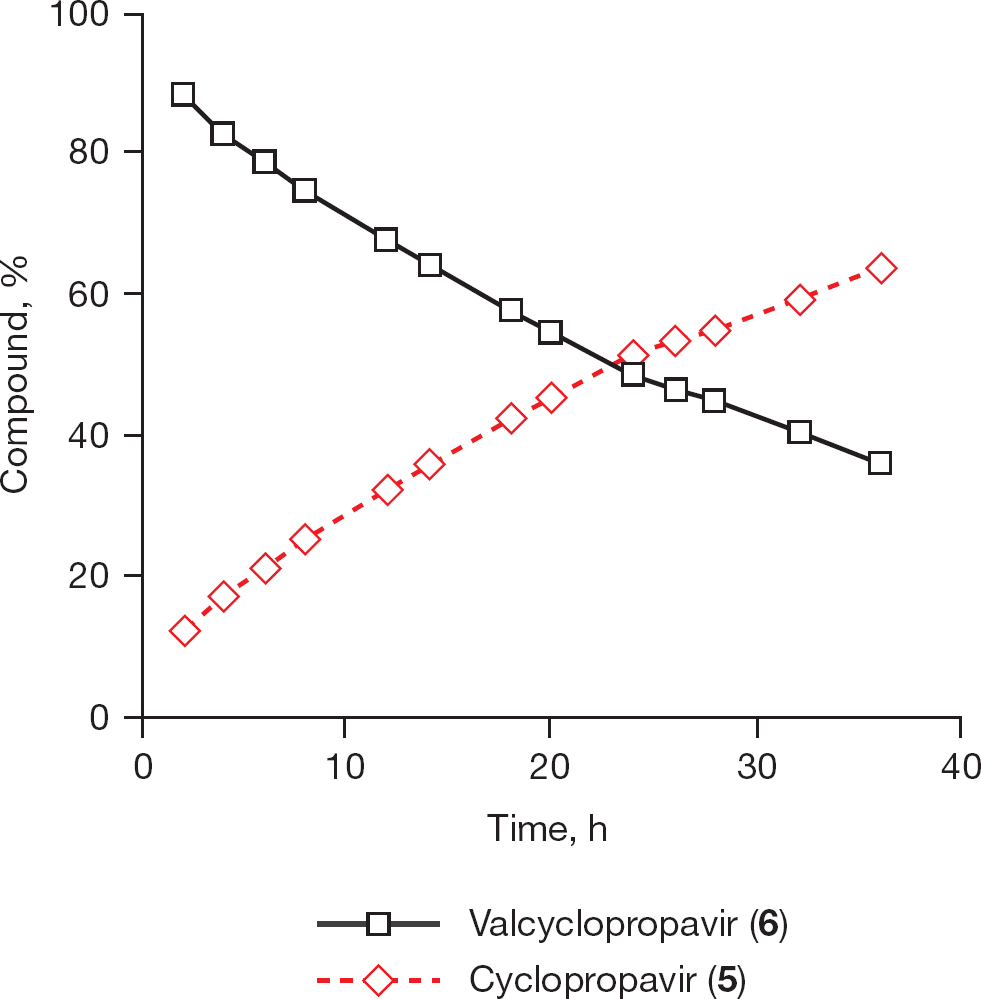
Kinetics of hydrolysis of prodrug (
Antiviral assays
Plaque reduction assays in human foreskin fibroblasts were performed with HCMV Towne and AD169 strains as previously described [14,15].
Stability of the prodrug valcyclopropavir (6) in mouse plasma and whole blood
Male Swiss–Webster mice (Taconic Farms, Germantown, PA, USA) were anaesthetized and blood was collected (1 ml per animal) by cardiac puncture into Monoject® collection tubes (number 8881-311149; Tyco Healthcare, Mansfield, MA, USA), containing an anticoagulant (0.04 ml of 7.5% EDTA[K3]). The blood was cooled in ice and used immediately for plasma preparation and stability studies. For plasma preparation, half of the collected blood was centrifuged for 10 min at 1,100 g in a refrigerated centrifuge (4°C). For stability at 0°C, ice cooled blood and plasma samples were spiked with a 10 mM solution of the prodrug (
Bioavailability of cyclopropavir (5) after intravenous and oral dosing of the prodrug valcyclopropavir (6) to mice
Dosing and blood sampling of mice
Prodrug (
Analytical methods
Preparation of standard solutions and spiked plasma calibration samples
Six standard solutions containing a mixture of prodrug (
Preparation and extraction of plasma samples from dosed mice
The plasma samples were thawed on ice, and aliquots (250 μl) were immediately transferred to microfuge tubes and deproteinated by addition of acetonitrile (250 μl). The samples were further processed as described above for the spiked plasma standards.
HPLC method
The HPLC analysis was performed on a Varian ProStar® HPLC System (Varian Inc., Palo Alto, CA, USA) consisting of two pumps, a variable wavelength detector, an autosampler and a desktop computer for HPLC control, data acquisition and data analysis. The data analysis software was the Varian® Star Chromatography Workstation (version 6.0). Compound elution was performed on a 4.6×150 mm Ace 5 C18 HPLC column (Advanced Chromatography Technologies, Aberdeen, Scotland, UK). The mobile phase consisted of reservoir A, containing 100% water with 0.1% trifluoroacetic acid, and reservoir B, containing 100% methanol. Each solvent was filtered through a 0.45 μm nylon filter and degassed before use. Elution from the column was at a flow rate of 1 ml/min using the following gradient programme: a linear gradient of 0% B to 30% B from 0 to 18 min, followed by a linear gradient from 30% to 100% B for 1 min by a hold at 100% B for 2 min, followed by a linear gradient from 100% to 0% B for 1 min, and then equilibration by a hold at 0% B for 5 min. Compound elution was monitored by UV absorbance at 274 nm. Total analysis time was 27 min per sample. Retention times were 9.8 min for cyclopropavir (
Data analyses
Peak areas from HPLC analysis of plasma samples were measured and concentrations of cyclopropavir (
Results
Antiviral activity
Prodrug (
Anti-HCMV activity of valcyclopropavir (

Efficacy and cytotoxicity in human foreskin fibroblasts (HFFs) as determined by plaque reduction assay.
Visual cytotoxicity in uninfected HFFs used in plaque reduction assays.
Cytotoxicity by neutral red assay.
Results from single assays.
The 50% effective concentration (EC50) values for cyclopropavir (
Conversion of valcyclopropavir (6) to cyclopropavir (5) in vitro in mouse plasma and whole blood
The prodrug valcyclopropavir (
Conversion of valcyclopropavir (

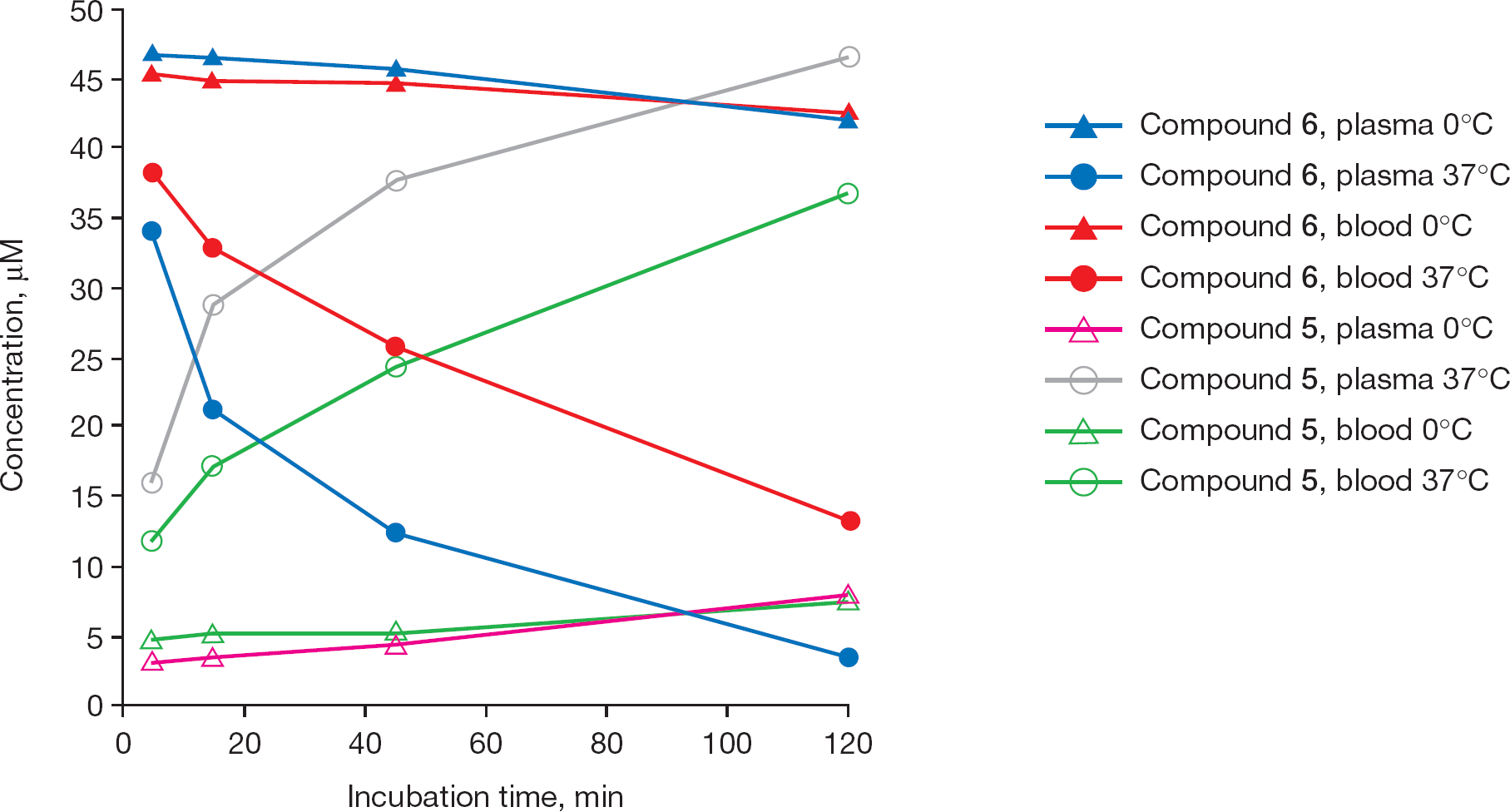
Conversion of prodrug (
Intravenous dosing
The average plasma concentration of the prodrug (
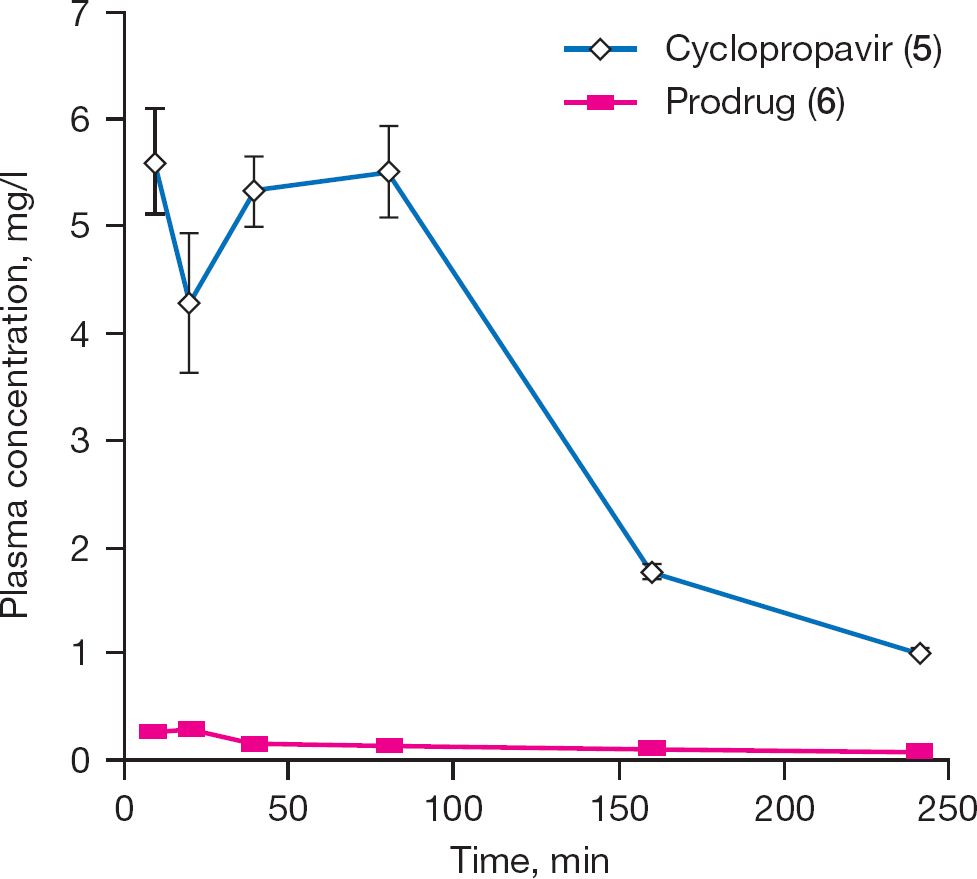
Plasma concentrations of cyclopropavir (
Prodrug (6)
The plasma concentration data for prodrug (
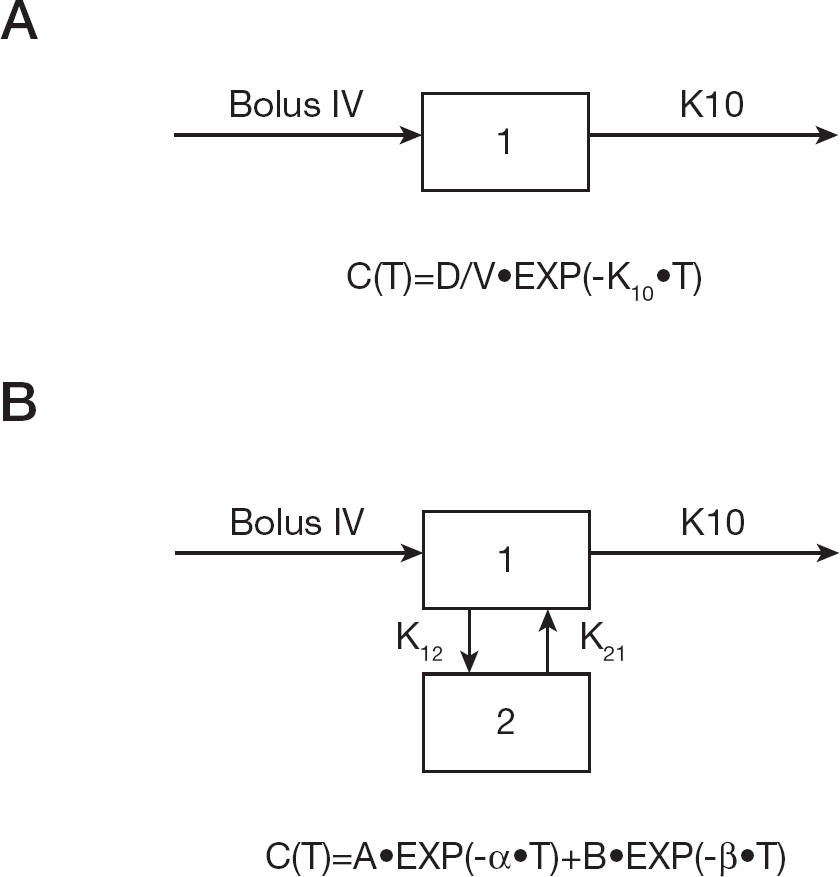
Single and two compartment bolus intravenous first order elimination models showing plasma concentration data for prodrug (
The best fit curve is presented in Figure 6A and the best fit pharmacokinetic parameters are presented in Table 3. Analysis of the plasma concentration profile suggested that the prodrug (
Single compartment first order elimination pharmacokinetic parameters for prodrug (
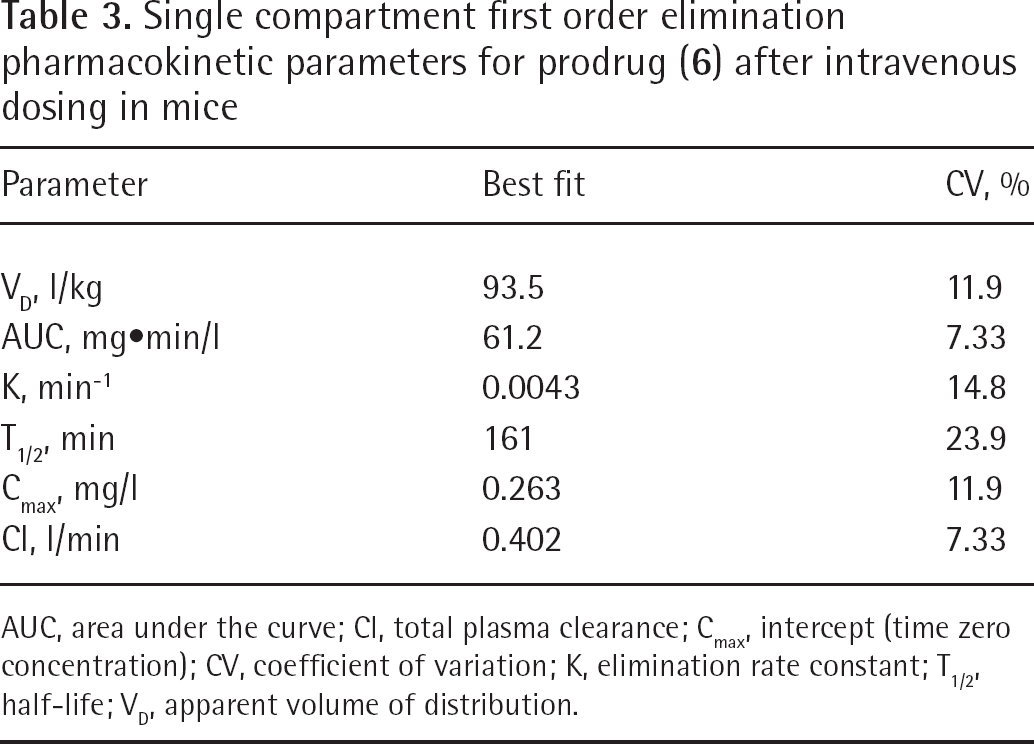
AUC, area under the curve; Cl, total plasma clearance; Cmax, intercept (time zero concentration); CV, coefficient of variation; K, elimination rate constant; T1/2, half-life; VD, apparent volume of distribution.
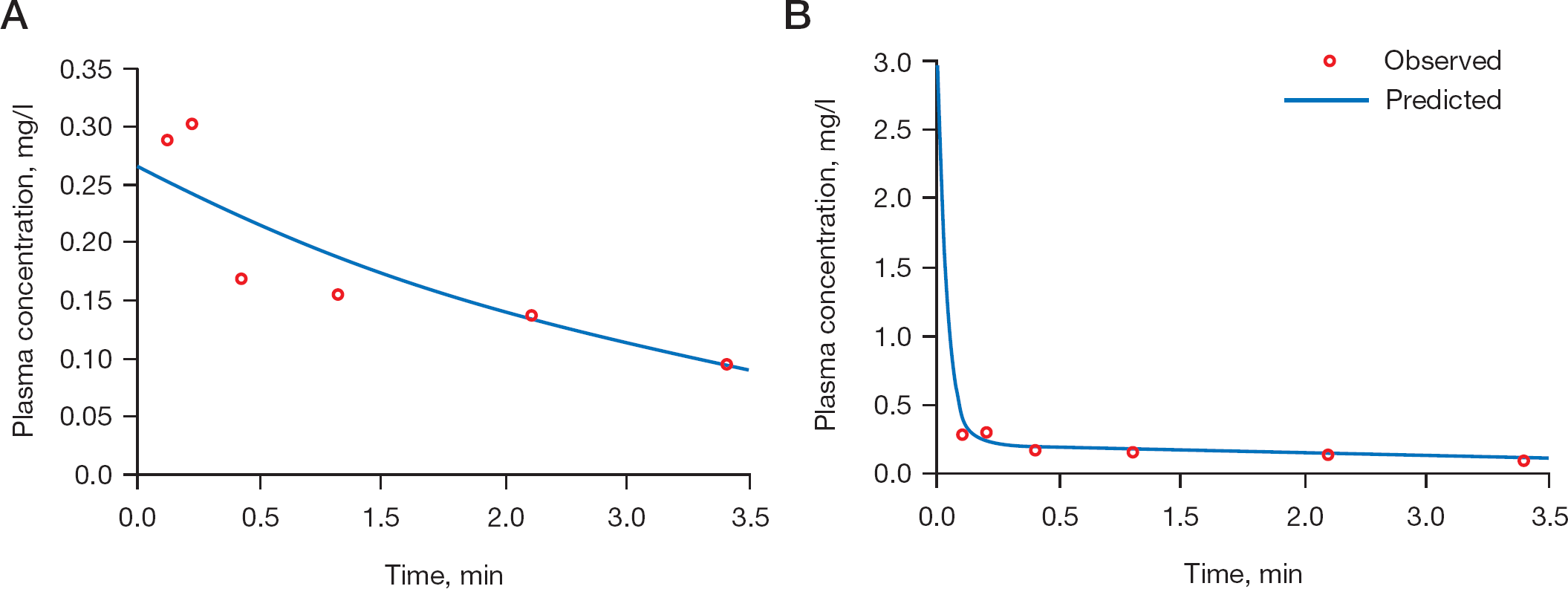
Pharmacokinetic modelling of the experimental data for prodrug (
Cyclopropavir (5)
The plasma concentration of cyclopropavir (
Two compartment first order distribution and elimination pharmacokinetic parameters for prodrug (
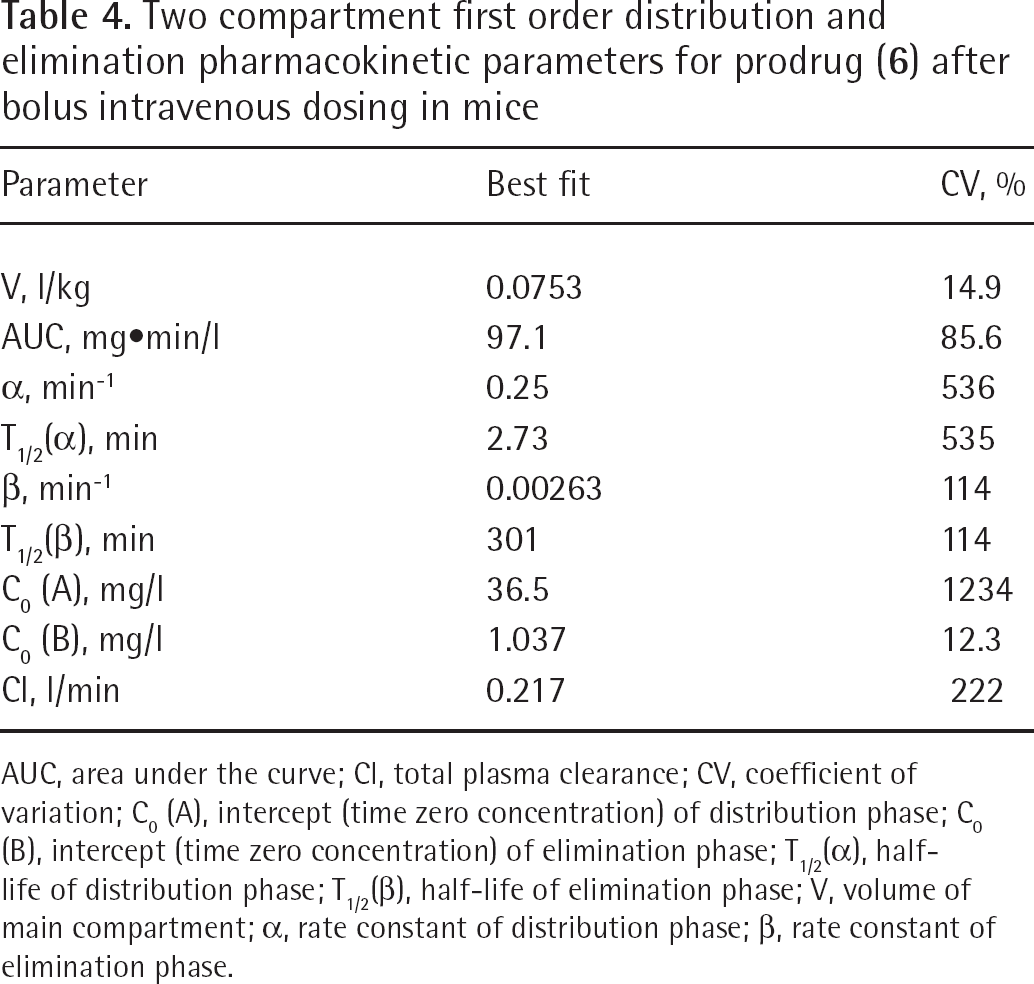
AUC, area under the curve; Cl, total plasma clearance; CV, coefficient of variation; C0 (A), intercept (time zero concentration) of distribution phase; C0 (B), intercept (time zero concentration) of elimination phase; T1/2(α), half-life of distribution phase; T1/2(β), half-life of elimination phase; V, volume of main compartment; α, rate constant of distribution phase; β, rate constant of elimination phase.
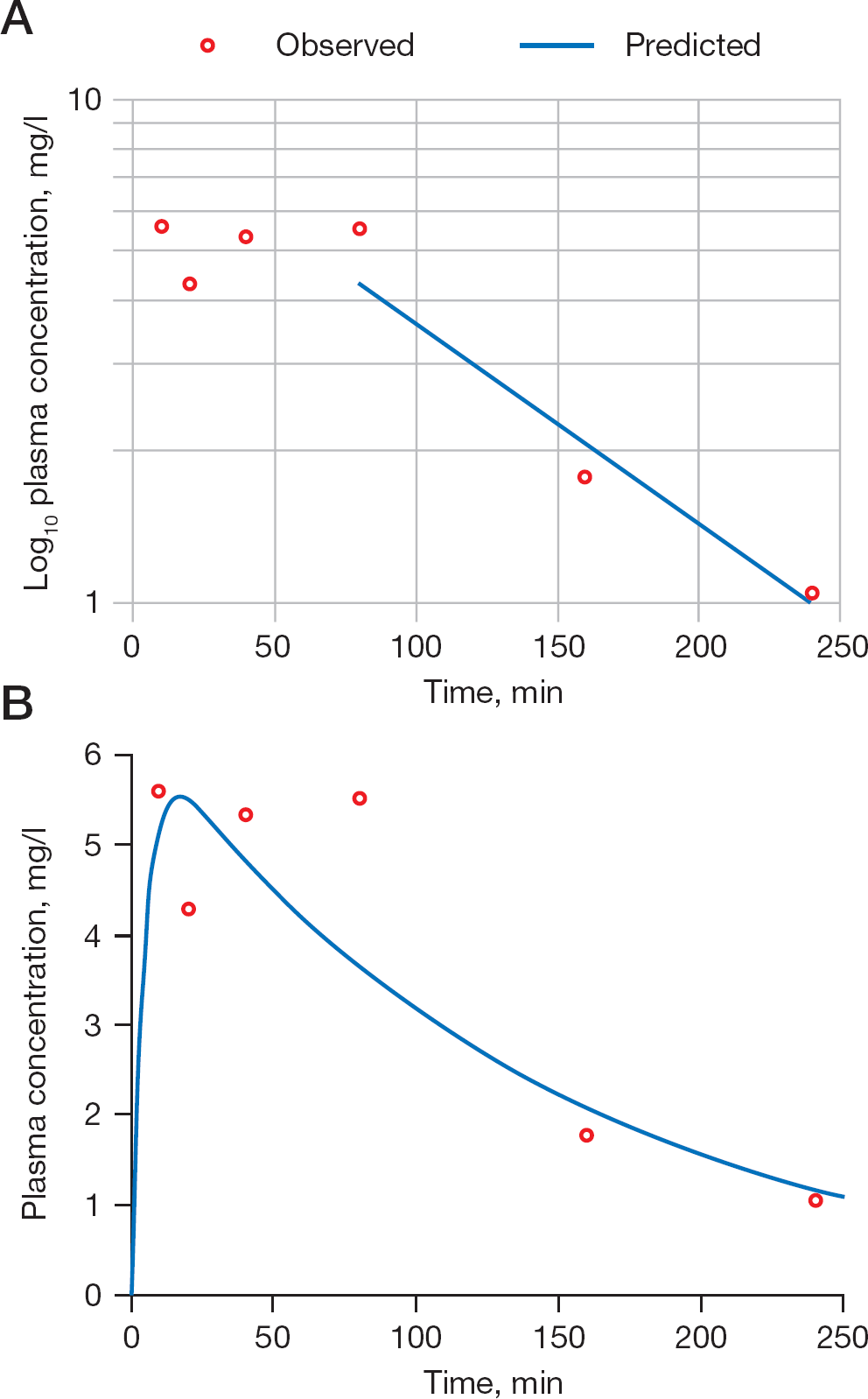
Pharmacokinetic modelling of the experimental data for cyclopropavir (
The time dependence of the cyclopropavir (
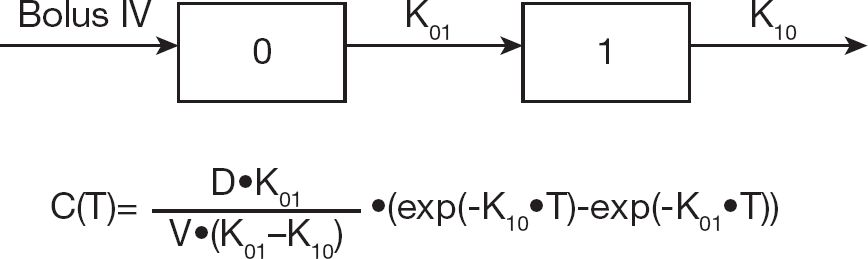
Two compartment model showing time dependence of the cyclopropavir (
The best fit graph and corresponding parameters are presented in Figure 7B and Table 5. The pharmacokinetic parameters obtained by this analysis are in good overall agreement with the corresponding pharmacokinetic parameters, obtained by the single compartment method (Table 6). However, because the elimination of the prodrug (
Two compartment analysis of cyclopropavir (
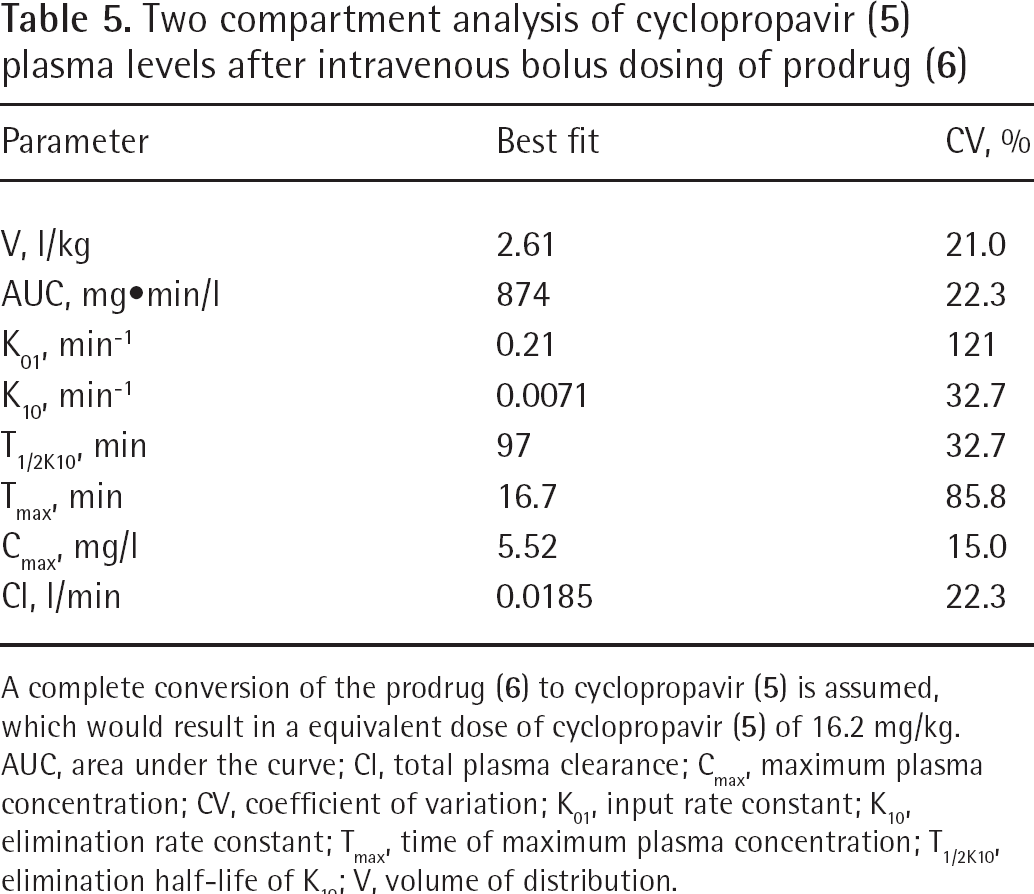
A complete conversion of the prodrug (
Single compartment first order elimination pharmacokinetic parameters for cyclopropavir (

The last three data points were used to model the elimination rate constant. In this analysis a complete conversion of the prodrug (
Oral dosing
The results of the plasma sample analysis after a single oral dose of 49.2 mg of prodrug (
Experimental and model-predicted concentrations of cyclopropavir (
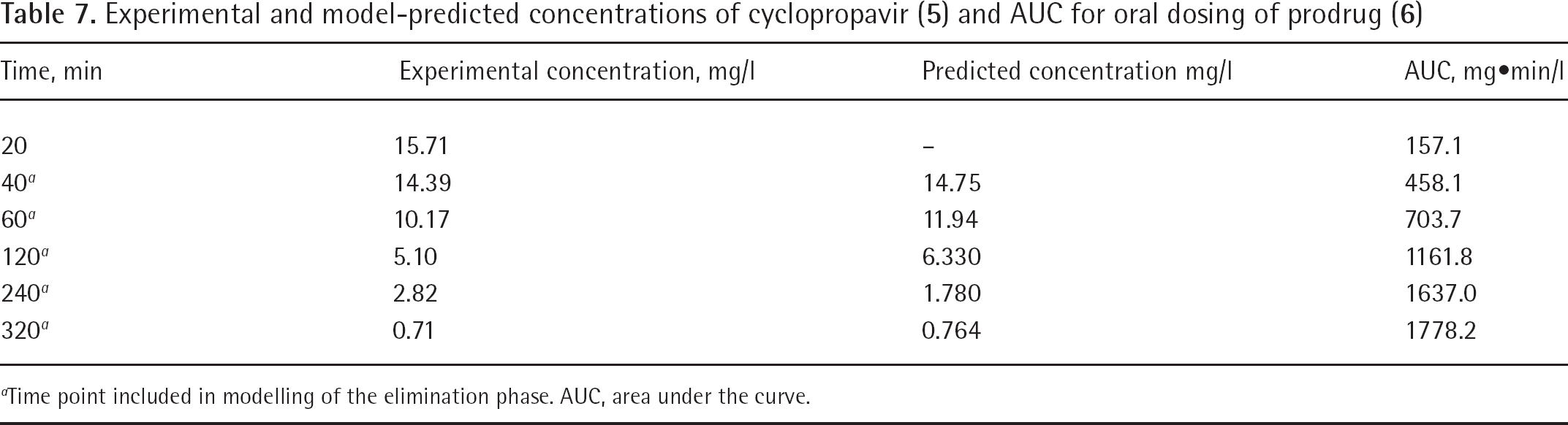
Time point included in modelling of the elimination phase. AUC, area under the curve.
Pharmacokinetic parameters of cyclopropavir (
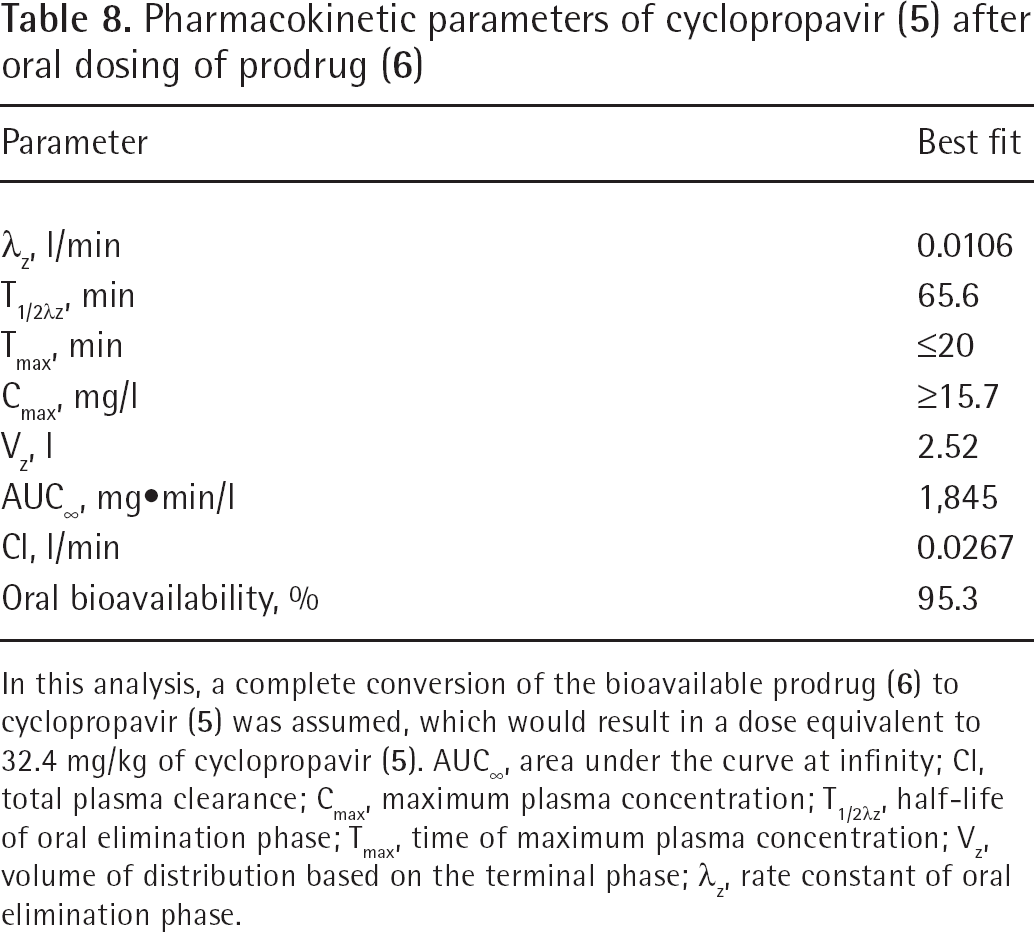
In this analysis, a complete conversion of the bioavailable prodrug (
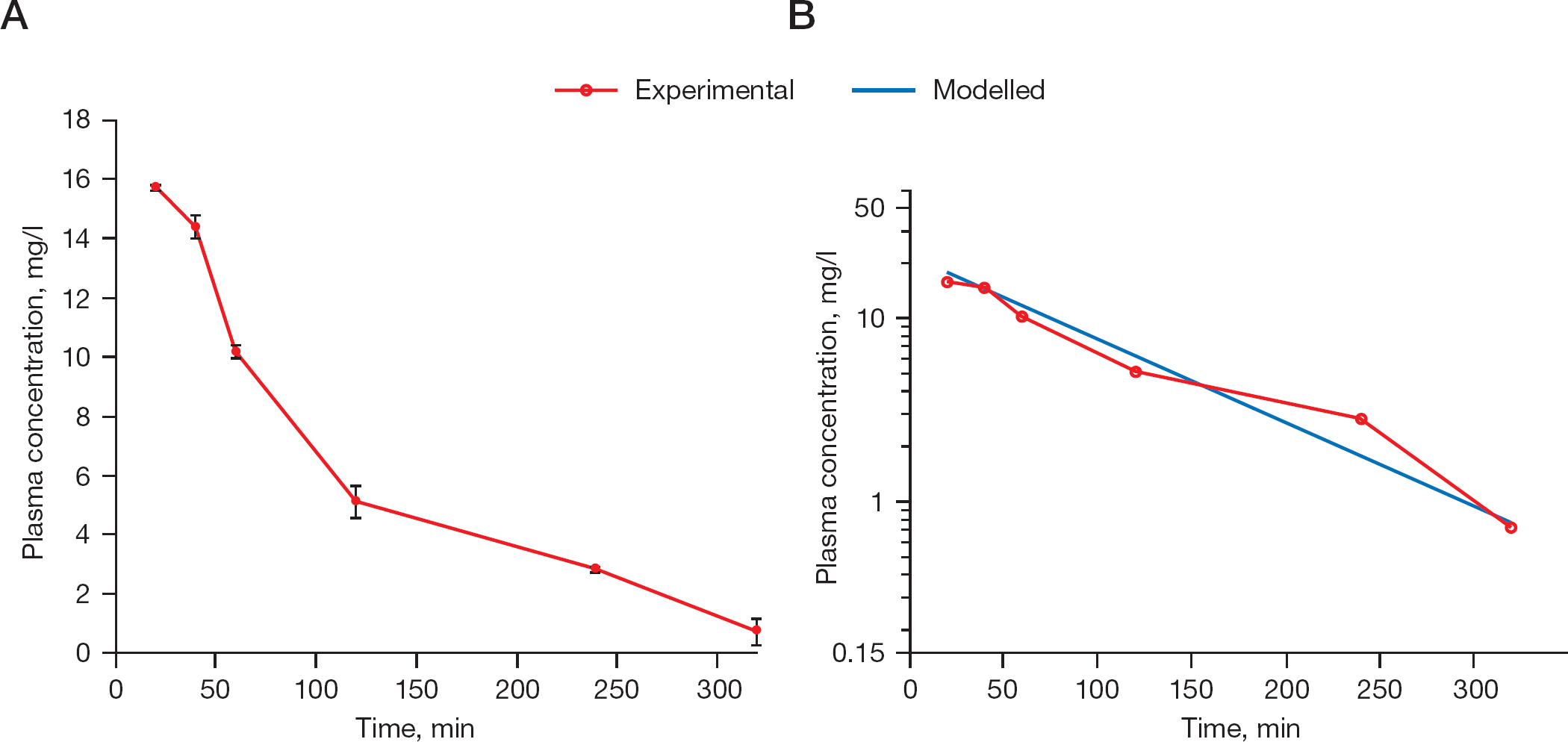
Plot of plasma concentrations of cyclopropavir (
Discussion
Chemistry
The previously described [17] tetrahydropyranyl (THP) cyclopropavir (
As expected, prodrug (
Pharmacokinetic studies
Prodrug (
Footnotes
Acknowledgements
The work described herein was supported by SBIR grant 5 R44 AI054135, contract NO1-AI30049 and research grant RO1-CA32779 from the National Institutes of Health, Bethesda, MD, USA. We thank LM Hrihorczuk (Central Instrumentation Facility, Department of Chemistry, Wayne State University, Detroit, MI, USA) for mass spectra, Kathy Borysko (University of Michigan, Ann Arbor, MI, USA) for plaque assays, and Sofya Dvoskin and George Wright (GLSynthesis, Worcester, MA, USA) for valuable assistance in bioavailability studies.
The authors declare no competing interests.