
Abstract
Background
Cobicistat (COBI), a CYP3A inhibitor, is a pharmacokinetic enhancer that increases exposures of the HIV protease inhibitors (PIs) atazanavir (ATV) and darunavir (DRV). The potential drug interaction between COBI-boosted PIs and hormonal contraceptives, which are substrates of intestinal efflux transporters and extensively metabolized by CYP enzymes, glucuronidation and sulfation, was evaluated.
Methods
This was a Phase I, open-label, two cohort (n=18/cohort), fixed-sequence study in healthy females that evaluated the drug–drug interaction (DDI) between multiple-dose ATV+COBI or DRV+COBI and single-dose drospirenone/ethinyl estradiol (EE). DDIs were evaluated using 90% confidence intervals of the geometric least-squares mean ratios of the test (drospirenone/EE+boosted PI) versus reference (drospirenone/EE) using lack of DDI boundaries of 70–143%. Safety was assessed throughout the study.
Results
29/36 participants completed the study. Relative to drospirenone/EE alone, drospirenone area under the plasma concentration versus time curve extrapolated to infinity (AUCinf) was 1.6-fold and 2.3-fold higher, and maximum observed plasma concentration (Cmax) was unaltered, upon coadministration with DRV+COBI and ATV+COBI, respectively. EE AUCinf decreased 30% with drospirenone/EE + DRV+COBI and was unchanged with ATV+COBI + drospirenone/EE, relative to drospirenone/EE alone. Study treatments were generally well tolerated. The majority of adverse events were mild and consistent with known safety profiles of the compounds.
Conclusions
Consistent with COBI-mediated CYP3A inhibition, drospirenone exposure increased following coadministration with COBI-containing regimens, with a greater increase with ATV+COBI. Thus, clinical monitoring for drospirenone-associated hyperkalaemia is recommended with DRV+COBI and ATV+COBI should not be used with drospirenone. Lower EE exposure with DRV+COBI may be attributed to inductive effects of DRV on CYP enzymes and/or intestinal efflux transporters (that is, P-gp) involved in EE disposition.
Introduction
Women comprise 50% of the global adult population (≥15 years and older) living with HIV. AIDS-related illnesses remain the leading cause of death for women of reproductive age (15–44 years) [1,2]. Hormonal contraceptives are used worldwide and are highly effective at preventing unintended pregnancies [3]. The concomitant use of hormonal contraception and antiretroviral (ARV) medications is increasingly common. Current US ARV guidelines recommend that ‘effective and appropriate’ options for contraception be offered to all women to reduce the risk of unintended pregnancies, and the World Health Organization recognizes access to reproductive health and contraceptive choice as a fundamental right that is essential to care for women and girls living with HIV [4,5].
Hormonal contraceptives are metabolized by cytochrome P450 (CYP) enzymes (that is, CYP3A4 and CYP2C9), glucuronidation and sulfation. Their disposition may also be influenced by efflux transporters such as P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) [6]. Drug–drug interactions (DDIs) between hormonal contraceptives and ARVs are well documented in the literature [7–12] and are of significance for certain agents, including ritonavir (RTV)-boosted protease inhibitors (PIs) and some non-nucleoside reverse transcriptase inhibitors (NNRTIs) as many are inhibitors or inducers of these metabolic and/or transport pathways [13,14].
The most widely prescribed combined hormonal oral contraceptives include a progestin (for example, norgestimate, norethindrone, drospirenone) and an estrogen component (for example, ethinyl estradiol [EE]), and both undergo extensive ‘first pass’ metabolism [15]. Drospirenone, a commonly used progestin, is a spironolactone analogue with antimineralocorticoid and antiandrogenic activity. It is extensively metabolized and the two main metabolites include the acid form of drospirenone and 4,5-dihydrodrospirenone-3-sulfate, which are both pharmacologically inactive [16]. It is a moderately sensitive CYP3A substrate, as evidenced by 2.68-fold higher exposure upon coadministration of drospirenone/EE and the strong CYP3A inhibitor ketoconazole [17]. The role of transporters in the disposition of drospirenone is unclear. Drospirenone is not a perpetrator of clinically relevant drug interactions in vivo [16].
EE undergoes extensive first-pass metabolism (both gut and hepatic) via multiple biotransformation pathways and has an average oral bioavailability of approximately 50% (range 20–65%) [6,16,18]. It is metabolized by CYPs, primarily CYP3A and to a minor extent by CYP2C9, and undergoes glucuronidation by UDP-glucuronosyltransferase 1A1 (UGT1A1) and sulfation by sulfotransferase 1E1 [6,19]. Efflux transporters such as P-gp and BCRP may also affect the disposition of EE [6,20]. Clinical studies have demonstrated that EE is a weak inhibitor of CYP3A and weak to moderate inhibitor of CYP2C19 and CYP1A2. Decreased plasma concentrations of certain drugs have also been reported when coadministered with EE, possibly due to EE-mediated induction of glucuronidation [16,21,22].
PI-based regimens are an important therapy option for people living with HIV due to their virological potency, high barrier to resistance and robustness of response in treatment-naive individuals with HIV. Although they are no longer recommended as first-line options for most people with HIV, they remain a favourable option for those with variable adherence or when initiation of early antiretroviral therapy is necessary (before results of resistance testing is available) [4]. Boosted DRV-based regimens also continue to be a key therapy for treatment-experienced patients [23]. Darunavir (DRV) and atazanavir (ATV) are primarily metabolized by CYP3A. To increase their systemic exposure, DRV and ATV are coadministered with the strong CYP3A inhibitor cobicistat (COBI) [24]. COBI-boosted DRV and ATV can perpetrate enzyme and transporter-mediated drug interactions. Both ATV+COBI and DRV+COBI are inhibitors of various enzymes and transporters, including CYP3A, CYP2D6, P-gp, the organic anion transporting polypeptides OATP 1B1/1B3, the organic cation transporter 2 (OCT2) and multidrug and toxin extrusion protein 1 (MATE1) [8,24–26]. Additionally, ATV also inhibits UGT1A1 and DRV may induce CYP metabolism and intestinal efflux transporters via PXR agonism [7,25,27–29]. Coadministration of ATV+COBI or DRV+COBI with substrates of these enzymes and/or transporters can result in complex drug–drug interactions that may warrant further evaluation.
Currently no clinical data are available to inform recommendations for the use of hormonal contraceptives with ATV+COBI and DRV+COBI. The objective of this study was to evaluate the pharmacokinetics (PK) of drospirenone and EE, a representative hormonal contraceptive, when co-administered with DRV+COBI or ATV+COBI in healthy women. Since drospirenone and EE were not expected to alter the PK of DRV, ATV or COBI, this study evaluated the effect of multiple doses of DRV+COBI and ATV+COBI on the PK of single doses of drospirenone/EE (a one-way interaction).
Methods
This was a Phase I, fixed-sequence, open-label, multiple-cohort, single-centre study conducted at PPD Development, LP (Austin, TX, USA). Eligible participants were healthy, non-pregnant, non-lactating, premenopausal female participants of 18 to 45 years of age with a body mass index (BMI) between 19 and 30 kg/m2. The study protocol and informed consent were approved by the study centre's Institutional Review Board, and participants provided written consent before study participation.
Major inclusion criteria included healthy participants based on medical history/physical examinations/laboratory evaluations, normal 12-lead electrocardiogram, normal renal function (creatinine clearance ≥90 ml/min), no evidence of HIV, HBV or HCV infection, non-smoker, not currently taking a hormonal contraceptive and willing to enrol in the study and receive single doses of drospirenone/EE. Exclusion criteria included plasma and blood donation within 7 and 56 days of study entry, respectively, active medical illness, use of prescription drugs within 28 days of study drug dosing (except vitamins, acetaminophen and/or ibu-profen), receipt of any hormonal contraception within 28 days prior to study dosing, receipt of medroxyprogesterone acetate (Depo-Provera) injection within 9 months of screening, use of an etonogestrel releasing implant (Implanon) within 28 days of screening, and current use of a progesterone releasing intra-uterine device (IUD).
Eligible participants were enrolled into one of two cohorts (Figure 1) where the drug–drug interaction (DDI) potential between DRV+COBI (800 mg + 150 mg; Cohort 1) or ATV+COBI (300 mg + 150 mg; Cohort 2) and single dose OC drospirenone/EE (drospirenone 3 mg/EE 0.02 mg) was evaluated. Each cohort received three treatments. A single dose of drospirenone/EE was administered on day 1 in both cohorts. In both cohorts, multiple daily doses of boosted-PIs were administered to steady-state based on historical data [30,31]. DRV 800 mg + COBI 150 mg were administered alone for 12 days (study days 5 to 16) in cohort 1, followed by coadministration with a single dose of drospirenone/EE (study day 17). In cohort 2, ATV 300 mg + COBI 150 mg were administered alone for 10 days (study days 5 to 14) followed by coadministration with single dose drospirenone/EE (study day 15). All study treatments were administered at approximately the same time each day in the morning with approximately 240 ml of water following an overnight fast (no food or drink, except water, for at least 10 h) and within 5 min of completion of a standardized moderate fat breakfast (approximately 600 kcal and 27% fat). On PK assessment days, food intake was restricted until after the 4-h blood sample was collected, and water intake was restricted 1 h before, and 2 h after dosing of study drug.

Study design
Safety assessments
Safety was evaluated by assessment of clinical laboratory tests (haematology profile, chemistry profile and urinalysis), physical examinations, vital signs, serum pregnancy tests, and review of concomitant medications performed at screening, at baseline (day before first study dose), on days before PK blood sampling, and at various times during the study. Participants were monitored for adverse events (AEs) throughout the study and follow-up. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA); AEs and lab abnormalities were graded according to the Gilead Grading Scale for Severity of Adverse Events and Laboratory Abnormalities. AEs were graded 1, 2, 3 or 4, based on severity, which were defined as mild, moderate, severe or life-threatening, respectively.
Pharmacokinetic evaluation
Serial blood samples were collected relative to study drug administration on study day 1 (all participants) and study day 17 (cohort 1) or study day 15 (cohort 2) as follows: predose (≤5 min prior to dosing), 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48, 72 and 96 h post-dose.
Timing of blood samples was based on known concentration-time profiles of each drug to accurately assess their PK. Blood samples were collected in a Vacutainer1 Plus plastic whole blood tube (Becton Dickinson, Franklin Lakes, NJ, USA) containing anticoagulant (spray-dried K2 ethylenediaminetetraacetic acid [EDTA]) and inverted several times to mix the blood and the anticoagulant. Tubes were kept on ice for 30 min and centrifuged for 10 min at 1,000 relative centrifugal force (RCF) in a refrigerated centrifuge set at 4°C to harvest plasma. Plasma samples were frozen at −70°C until analysis.
Bioanalytical procedures
Concentrations of EE, COBI, DRV, ATV and drospirenone in human plasma samples were determined using fully validated high-performance liquid chromatography-tandem mass spectroscopy (LC/MS/MS) bioanalytical methods. All samples were analysed in the timeframe supported by frozen stability storage data. The assays for EE, COBI, DRV and ATV were all performed and validated by QPS, LLC (Newark, DE, USA). The assays for drospirenone were performed and validated by Syneos Health (Princeton, NJ, USA).
Sample analyses for plasma concentrations of EE, COBI, DRV and ATV were performed as previously described [26,30,32]. The appropriate dilution factor was applied to the result to generate a reported value. Concentrations below the calibrated ranges of the methods were reported as BQL (below the quantification limit). For drospirenone determination, 100 μl of human plasma was spiked with drospirenone-d4, and the sample was then processed by a protein precipitation extraction procedure. After evaporation of the organic solvent, an aliquot of the reconstituted sample extract was injected onto the HPLC-MS/MS system. The calibrated range of the method was 0.25 to 100 pg/ml. All % coefficient of variation (CV) values were <6.89%, and all % relative error (RE) values were within ±6.8%, of 100%. Stability in frozen matrix was 58 days at −20°C to −70°C.
PK Analyses
PK parameters were estimated by non-compartmental methods, using linear up/log down method for area under the plasma concentration-time curve (AUC) estimation (WinNonlin software, version 6.4; Pharsight Corporation, Princeton, NJ, USA). PK parameters included the area under the plasma concentration versus time curve extrapolated to infinity (AUCinf: EE and drosperinone), area under the plasma concentration versus time curve from time zero to the last quantifiable concentration (AUClast), area under the plasma concentration versus time curve over the dosing interval (AUCtau: ATV, DRV and COBI), maximum observed plasma concentration (Cmax), time to reach maximum concentration (Tmax), observed plasma concentration at the end of the dosing interval (Ctau: ATV, DRV and COBI), last quantifiable concentration (Clast), time of Clast (Tlast), and terminal elimination half-life (T1/2). To avoid bias in the estimation of the terminal elimination rate constant, samples that were below the lower limit of quantitation after the first quantifiable time point were treated as missing data.
Statistical analyses
The sample size of 16 participants per cohort was estimated on the basis of the following assumptions: the estimated two-sided 90% CI of the geometric least-squares means (GLSM) ratio of test versus reference treatments with regards to AUC and Cmax will be within 0.70, 1.43 with ≥90% probability if the true GLSM ratio was 1.0. This is assuming a standard deviation (sd) of differences of no more than 0.400 on a natural logarithm scale for the most variable PK parameter, EE Cmax. The ‘lack of DDI’ boundaries in this study were informed by results of previous DDI studies between approved drugs and oral contraceptives that showed no clinically meaningful change in exposure of the oral contraceptive [20,33ȃ36]. A total of 18 participants per cohort were enrolled which included a 10% overage.
An analysis of variance (ANOVA) using a mixed-effects model with treatment as a fixed effect and participant as a random effect was fitted to the natural logarithmic transformation of PK parameters for each analyte. Two-sided 90% confidence intervals (CIs) were calculated for the ratio of GLSM of primary PK parameters between test (drospirenone/EE) versus reference (ATV+COBI or DRV+COBI with drospirenone/EE) treatments for each analyte and compared against lack of PK DDI bounds of 70% to 143%, inclusive, for primary PK parameters of drospirenone and EE (AUCinf, AUClast and Cmax).
Results
A total of 36 female participants (18/cohort) were enrolled and 29 subjects (80.6%) completed the study. The majority of participants were White (52.8%) or Black or African American (44.4%) with a mean age of 31 years (range: 18–44 years). The mean BMI at baseline was 26.1 kg/m2. The mean estimated glomerular filtration rate calculated using the Cockcroft–Gault equation (eGFRCG) at baseline was 125.8 ml/min. Seven participants (19.4%) discontinued study drug and the study due to Grade 1 maculopapular rash (cohort 1: three participants [16.7%]; cohort 2: four participants [22.2%]), a known AE associated with these boosted PIs [7,8]. All the rash events were assessed by the investigator as related to study treatment and resolved following treatment with topical and/or oral antihistamines or corticosteroids (that is, hydrocortisone 1% cream, or diphenhydramine or methylprednisolone given orally).
Safety
Study drug treatments were generally well tolerated. There were no Grade 3 or 4 AEs. Overall 32 participants had an AE, of which 13 were in cohort 1 and 19 were in cohort 2. Of the 32 participants who experienced any AEs, 4 participants in cohort 1 and 16 participants in cohort 2 experienced AEs that were considered by the investigator to be drug related. The most frequently reported AEs (with some participants having had multiple AEs) considered related to study drug in cohort 1 were maculopapular rash (three participants [16.7%]) while receiving DRV+COBI. In cohort 2, hyperbilirubinaemia (15 participants [83.3%]), ocular icterus and maculopapular rash (both 5 participants [27.8%]) and diarrhoea (2 participants [11.1%]) were the most frequently reported AEs considered related to study drug while receiving ATV+COBI. All of these AEs were Grade 1 in severity except for Grade 2 angioedema reported for one participant in cohort 2.
There were no clinically relevant changes in median laboratory values throughout the study. In cohort 1, all laboratory abnormalities were Grade 1 or 2 in severity. In cohort 2, Grade 3 hyperbilirubinaemia was reported for 11 participants (61.1%) while receiving ATV+COBI and 8 participants (57.1%) while receiving ATV+COBI plus drospirenone/EE; Grade 4 hyperbilirubinaemia was reported for 4 participants (22.2%) while receiving ATV+COBI and 2 participants (14.3%) while receiving ATV+COBI plus drospirenone/EE. These results are consistent with the known safety profile of ATV and its inhibition of uridine 5′-diphospho-glucuronosyltransferase 1A1 (UGT1A1) [7].
There were increases in serum creatinine across both cohorts and corresponding decreases in eGFRCG while participants were receiving DRV+COBI (median creatinine increase from baseline of 0.15 mg/dl) or ATV+COBI (median creatinine increase from baseline of 0.10 mg/dl) that recovered to near baseline at day 21 and day 19, respectively. These findings are consistent with COBI-mediated inhibition of renal transporters involved in creatinine elimination (OCT2 and MATE1) [26]. No participant had a graded serum creatinine, serum phosphate and proteinuria abnormality during the study.
Pharmacokinetics
Drospirenone and EE PK parameters following administration of DRV+COBI + drospirenone/EE or drospirenone/EE alone (cohort 1)
The mean (
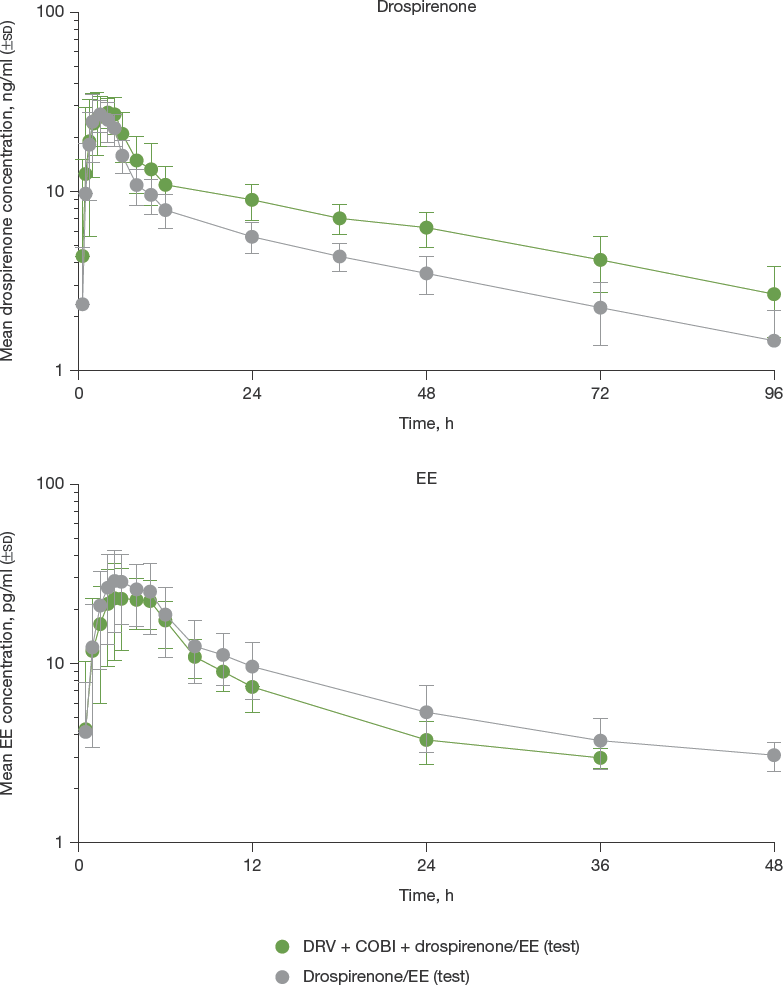
Mean (
Drospirenone and EE PK parameters following administration of DRV+COBI+ drospirenone/EE or drospirenone/EE alone in cohort 1

Terminal elimination half-life (T1/2) presented as median (Q1, Q3). AUCinf, area under the plasma concentration versus time curve extrapolated to infinity; CL/F, apparent clearance; Cmax, maximum observed plasma concentration; COBI, cobicistat; CV, coefficient of variation; DRV, darunavir; EE, ethinyl estradiol; GLSM, geometric least-squares mean; PK, pharmacokinetic.
Drospirenone and EE PK parameters following administration of ATV+COBI + drospirenone/EE or drospirenone/EE alone (cohort 2)
The mean (

Mean (
Drospirenone and EE PK parameters following administration of ATV+COBI+ drospirenone/EE or drospirenone/EE alone in cohort 2

Terminal elimination half-life (T1/2) presented as median (Q1, Q3). AUCinf, area under the plasma concentration versus time curve extrapolated to infinity; CL/F, apparent clearance; Cmax, maximum observed plasma concentration; COBI, cobicistat; CV, coefficient of variation; EE, ethinyl estradiol; GLSM, geometric least-squares mean; PK, pharmacokinetic.
The steady state PK parameters of DRV, ATV and COBI, as shown in Tables 3 and 4, were consistent with those observed previously [25].
DRV+COBI PK parameters following administration of DRV+COBI+ drospirenone/EE

AUCtau, area under the plasma concentration versus time curve over the dosing interval; Cmax, maximum observed plasma concentration; COBI, cobicistat; Ctau, observed plasma concentration at the end of the dosing interval; CV, coefficient of variation; DRV, darunavir; EE, ethinyl estradiol; PK, phrmacokinetic.
ATV+COBI PK parameters following administration of ATV+COBI+ drospirenone/EE

ATV, atazanavir; AUCtau, area under the plasma concentration versus time curve over the dosing interval; Cmax, maximum observed plasma concentration; COBI, cobicistat; Ctau, observed plasma concentration at the end of the dosing interval; CV, coefficient of variation; EE, ethinyl estradiol; PK, pharmacokinetic.
Discussion
Consistent with inhibition of CYP3A-mediated metabolism of drospirenone by COBI, this study demonstrated clinically significant increases in drospirenone exposures (AUCinf) of 58% and 130% in the presence of DRV+COBI or ATV+COBI, respectively. The reason for the difference in magnitude of increase in drospirenone exposure with DRV+COBI and ATV+COBI is unclear. The CYP3A inhibition potency of COBI is similar to ketoconazole [37,38], and the drospirenone AUC increase observed with ATV+COBI was similar to that demonstrated with drospirenone/EE and ketoconazole [17]. These data suggest that DRV may induce enzymes and/or transporters involved in the disposition of drospirenone. Drospirenone is an antimineralocorticoid with potassium-sparing properties [16,37]. As such, the use of strong CYP3A4 inhibitors (for example, azole antifungals, HIV/HCV protease inhibitors, clarithromycin) that increase drospirenone exposure (> twofold) and consequently increase the risk of hyperkalaemia is either recommended with clinical monitoring for hyperkalaemia (Tybost® Summary of Product Characteristics) or is contraindicated (Tybost® U.S. Prescribing information) [25,39]. The recommendation for the use of ATV+COBI with drospirenone-containing contraceptives should therefore be informed by local prescribing information for drospirenone/EE (Yaz®) or cobicistat (Tybost®).
Given the smaller (< twofold) increase in drospirenone exposure, it may be coadministered with DRV+COBI with clinical monitoring for hyperkalaemia, as recommended by the drospirenone/EE prescribing information [16].
Exposures of EE were numerically (18 to 22%) lower with ATV+COBI. The 90% CIs for the GLSM ratio. EE AUCinf and Cmax remained within the lack of DDI boundary when coadministered with ATV+COBI, indicating a lack of a clinically meaningful interaction. The mechanism for EE exposure reduction is unknown.
EE exposures (AUCinf) were 30% lower with DRV+COBI. EE is extensively metabolized by CYP enzymes (that is, CYP3A, CYP2C9), and intestinal efflux drug transporters (that is, P-gp/BCRP) may play a role in its absorption [6]. Reduction of EE exposure (44% decrease in AUC) was also observed following coadministration of norethindrone/EE with DRV/r and has been attributed to DRV/r-mediated induction of these pathways [27,28,30,40]. The magnitude of reduction in EE exposure observed in the current study with DRV+COBI was smaller and may be due to the lack of induction liability with COBI [8]. The efficacy of combined hormonal contraceptives is attributed largely to the progestin component rather than the estrogen component [41,42]. As such, the decrease in EE with DRV+COBI is not anticipated to compromise contraceptive efficacy. Comparisons of steady state PK parameters of DRV, ATV and COBI to historical data demonstrated that they were consistent with those observed previously.
All treatments were generally well tolerated when administered alone and in combination. Safety findings in this study were consistent with the known safety pro-files of the study drugs. Maculopapular rash, reported in seven participants (19.4%) who discontinued the study (and study drug), is a known adverse drug reaction for COBI, DRV and ATV; the frequency of this AE was similar to what has been reported for these boosted PIs [7,8]. Hyperbilirubinaemia, reported in participants receiving ATV+COBI, is a known adverse effect of ATV through its inhibitory effect on UGT enzymatic activity [43]. The incidence of hyperbilirubinaemia reported for participants while receiving ATV+COBI in cohort 2 (15 participants, 83.3%) are consistent with the prescribing information for ATV [7]. The changes in serum creatinine seen with the COBI-containing regimens are consistent with its known safety profile. Cobicistat inhibits renal tubular creatinine secretion via inhibition of OCT2 and MATE1, resulting in an increase in serum creatinine and a decrease in eGFRCG, without affecting actual glomerular filtration rate [26]. The increases in serum creatinine and corresponding decreases in eGFRCG for both cohorts recovered to near baseline 5 days after antiretroviral washout. There were no clinically relevant changes in serum creatinine or eGFRCG while participants were receiving drospirenone/EE alone, or drospirenone/EE plus the ARVs in both cohorts. There were no other clinically relevant findings in relation to renal safety.
In conclusion, the results of this study showed that treatment with the COBI-containing regimens DRV+COBI and ATV+COBI increased drospirenone exposures but did not alter EE exposures to a clinically relevant extent. Increased drospirenone exposures, such as those observed in the setting of coadministration with a strong CYP3A inhibitor, are associated with the risk of hyperkalaemia, and recommendations for use of drospirenone/EE with strong CYP3A inhibitors vary by geographic region [16,44]. In general, clinical monitoring of serum potassium concentrations is recommended when strong CYP3A inhibitors are coadministered with drospirenone/EE. Local prescribing information for drospirenone/EE (Yaz®) or cobicistat (Tybost®) should be consulted prior to coadministration with ATV+COBI or DRV+COBI. The steady state PK parameters of DRV, ATV and COBI were consistent with historical data. Study drug treatments were generally well tolerated and no new or unexpected safety findings in this short-term study in healthy participants.
Footnotes
All authors are employees of Gilead Sciences.