
Abstract
Background
Tenofovir disoproxil fumarate (TDF), the oral prodrug of tenofovir (TFV), is advocated in pregnancy for prevention of mother-to-child transmission (PMCT) with failure of hepatitis B immunoglobulin and vaccination. The pharmacokinetics of TDF monotherapy for PMCT-HBV is important if deployment is to emulate the success of multiple antiretrovirals (ARVs) for PMCT-HIV in resource-constrained settings.
Methods
This systematic review followed a protocol and is reported according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement (PRISMA) guidelines. We included studies that enrolled pregnant women who received oral TDF therapy as mono-therapy or in combination with other ARVs: irrespective of the reason for receiving the drug (for example, HIV, HBV or pre-exposure prophylaxis); and reported pharmacokinetics.
Results
The area under the concentration–time curve (AUC), maximum plasma concentrations (Cmax) and last measurable plasma concentration (Clast) of TFV were decreased in the second and third trimester compared with first trimester or post-partum. In none of the manuscripts was the non-pregnant HBV threshold of Cmax of 300 ng/ml reached, but the 50% effective concentration (EC50) of TFV is lower for treatment of HBV compared with HIV. The TFV concentration in breastfed infants was 0.03% of the recommended infant dose.
Conclusions
Most knowledge of pharmacokinetics of TFV in pregnancy results from studies on HIV involving multiple ARVs. Increased TFV clearance occurred in the second and third trimester when optimal TFV concentrations are required to maximize suppression of HBV in the window before birth. Dose or duration adjustments will be better conceptualized with concurrent analysis of the pharmacokinetics of TFV monotherapy and hepatitis B pharmacodynamics in pregnancy.
Introduction
Over 3% of the world's population are infected with HBV, which is the most significant risk factor for hepatocellular carcinoma. Mother-to-child transmission (MTCT) is the main route of acquisition [1–4] and the risk of infection is linear to maternal viral load at birth [5]. Perinatal infection occurs in 70–90% of babies born to women with hepatitis B e antigen-positive (HBeAg) HBV and 0–30% in those who are HBeAg-negative [1]. Even with optimal preventive strategies, hepatitis B immunoglobulins (HBIG) after birth in HBeAg-positive mothers in addition to HBV vaccination, MTCT occurs in an estimated 8–32% of cases [6,7]. Unfortunately, in low-income countries (LIC) where HBV is most prevalent, HBIG is usually not provided because of access (including homebirth), complexity of production and delivery, and short shelf life, cost, and the need for a cold chain [8].
Maternal tenofovir disoproxil fumarate (TDF) therapy is one strategy under consideration to reduce MTCT. Pregnant women could be treated with oral TDF during the course of their pregnancy and, if no further availability, it can be stopped 1 month post-partum. TDF is an oral prodrug of tenofovir (TFV), a nucleotide analogue that was developed and has been widely used as an antiretroviral drug against HIV and HBV. TDF has an oral bioavailability (F) estimated at 20–30%, and is formulation dependent [9]. Following absorption and distribution, TFV is converted intracellularly to its active anabolite tenofovir diphosphate (TDP). There is no efficacy threshold level for TFV in HIV, but thresholds for HBV that have been used included a maximum plasma concentration (Cmax) of 300 ng/ml and a half life (t 1/2 ) of 17 h (tested in non-pregnant males and females) [10]. The elimination of plasma or serum TFV is more rapid (t 1/2 12–16 h) than the bioactive intracellular TDP (t 1/2 87 h in peripheral blood mononuclear cells [PBMCs], and 96 h in hepatocytes) [11]. This t 1/2 of TFV in PBMCs is helpful since TFV is pharmacologically ‘forgiving’ in the context of poor adherence. TFV exhibits long-lasting anti-HBV activity in cell culture but has an in vitro 50% effective concentration (EC50) that is lower for treatment of HBV compared with HIV (0.03 ±0.02 μg/ml with continuous exposure) [12]. TDF is widely regarded as safe in pregnancy after extensive use in HIV-positive pregnant women [13].
Both the pharmacokinetic (PK) [14] and pharmacodynamic (PD) properties [15] of drugs may be affected by physiological changes of pregnancy and reduced exposure has been reported for antiretroviral drugs in pregnancy [14,16] and in lactation [17]. TDF suppression of HBV for prevention of MTCT starts in pregnancy which sets a constraint on the time frame available to reduce the viral load before birth. While a systematic review and meta-analysis supports high efficacy of TDF from 28 weeks given with HBIG and the birth dose to reduce MTCT; HBV DNA remains detectable in 2–38% of participants which is important because the threshold of HBV DNA load to prevent MTCT of HBV without HBIG (reality for resource limited settings) is unknown [6,18–20]. Rebound of HBV DNA to pre-treatment levels within 4–8 weeks occurs following short-course TDF before protective infant HBV antibody levels can be acquired [21]. This may be important in resource-constrained settings where breastfeeding remains the only source of infant nutrition and HBIG and HB birth dose administration are frequently not provided.
The objective of this systematic review was to summarize the PK of oral TDF for prevention of mother-to-child transmission (PMCT) of HBV and HIV in pregnant and lactating women. The PK of TDF monotherapy for PMCT-HBV may be important before widespread deployment in low-resource settings where optimal provision of HBIG and birth dose are problematic.
Methods
This systematic review followed a protocol and is reported according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement (PRISMA) guidelines.
Protocol and registration
This review was registered in advance in PROSPERO (International prospective register of systematic reviews). Registration number: CRD42018082352.
Eligibility criteria
We included studies that enrolled pregnant women who received oral TDF therapy as monotherapy or in combination with other antiretrovirals, irrespective of the reason for receiving the drug (for example, HIV, HBV or pre-exposure prophylaxis [PrEP]), and details of the reported PK. Both English and non-English-language studies were included.
Information sources
We searched for publications in Medline, Embase and Ovid Cochrane Central Register of Controlled Trials from 1980 to 15 November 2018. Controlled vocabulary supplemented with keywords was used to search for PK studies of TDF in pregnancy. A manual search of bibliographies of the included studies and relevant systematic reviews was conducted. We also contacted people in the field to make sure we did not miss unpublished papers. The last search was performed on 15 November 2018.
The complete search is listed in Additional file 1.
Study selection
Two independent reviewers screened in duplicate titles and abstracts for potential eligibility. Disagreements were reconciled by consensus or by a third reviewer. Articles that were selected were first screened for eligibility using the title and abstracts. Manuscripts were excluded during the process that did not meet our patient, intervention, control, outcome (PICO) objectives.
Data extraction
For each study, data extraction was done in duplicate using a standardized, pretested form. A third reviewer compared data and resolved inconsistencies by referring to the full text of the articles.
Data items
The exposure parameters used were those that were previously reported as being essential for dosing decision support in pregnancy, that is, area under the concentration–time curve (AUC, AUC∞ as total exposure, or within an interval, for example, in the case of daily dosing as AUC0–24), minimal (Ctrough) and maximal (Cmax) plasma concentrations [14]. Other distribution parameters collected were standard PK measures including: volume of distribution (Vd), fraction of drug bound and unbound to plasma proteins (fb and fub); and elimination parameters t 1/2 and clearance (CL).
Risk of bias assessment
Two reviewers assessed the risk of bias (that is, systematic error) independently using the Cochrane Risk of Bias assessment [22]. The quality of evidence (that is, certainty in the estimates) was evaluated using the Grading of Recommendations Assessment, Development, and Evaluation approach (GRADE) [23].
Criteria used to evaluate quality of evidence were risk of bias, indirectness (surrogate outcomes), imprecision (wide confidence intervals), inconsistency (heterogeneity) and publication bias [24].
Results
The initial search resulted in 668 citations of which 62 were duplicates. We eventually included 11 studies that were published between 2008 and 2018 (Table 1). Six studies informed about TDF PK during pregnancy [25–30] and five on drug concentrations of which three were after a single dose of drugs during childbirth [31–35]. The average weighted kappa for study selection was 0.71 (good, Kappa calculation in Additional file 1). The study selection process and reasons for exclusions are shown in Figure 1.
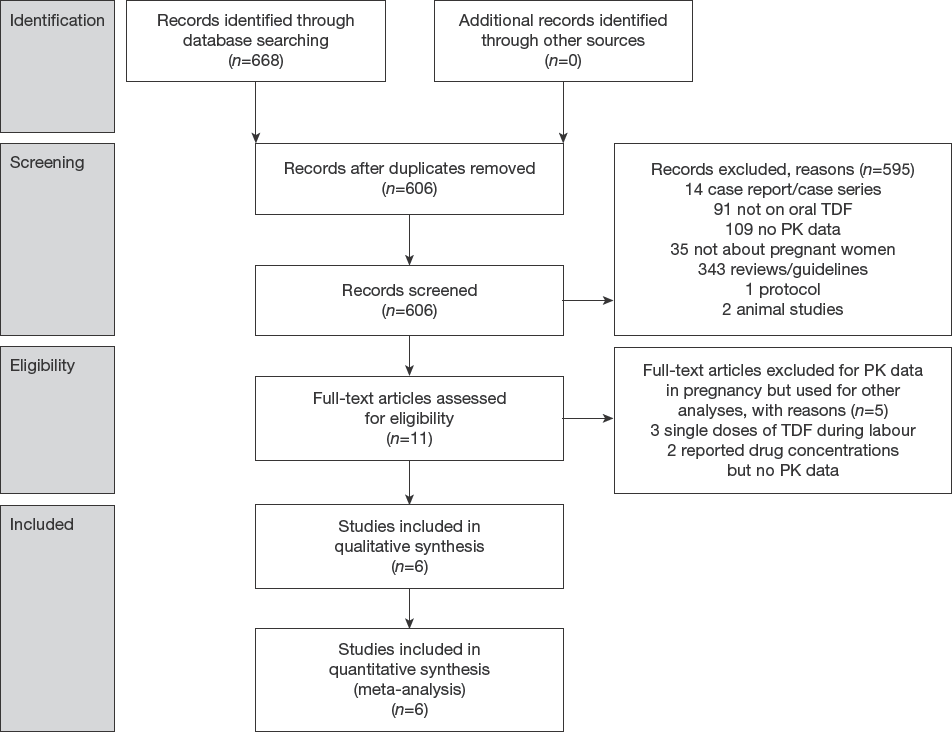
PRISMA flow diagram for PK studies on TDF in pregnancy
Summary of evidence
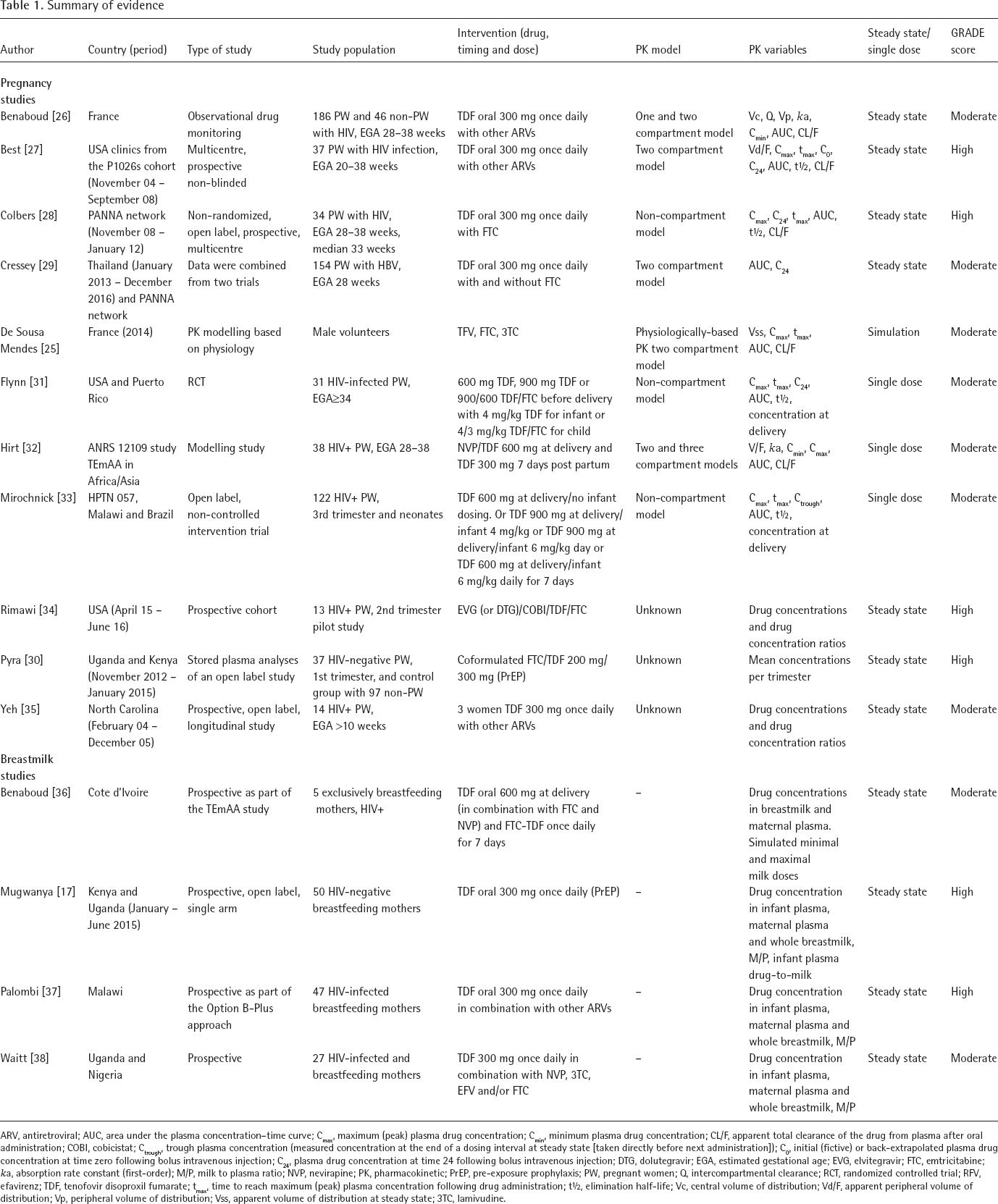
ARV, antiretroviral; AUC, area under the plasma concentration-time curve; Cmax, maximum (peak) plasma drug concentration; Cmin, minimum plasma drug concentration; CL/F, apparent total clearance of the drug from plasma after oral administration; COBI, cobicistat; Ctrough, trough plasma concentration (measured concentration at the end of a dosing interval at steady state [taken directly before next administration]); C0, initial (fictive) or back-extrapolated plasma drug concentration at time zero following bolus intravenous injection; C24, plasma drug concentration at time 24 following bolus intravenous injection; DTG, dolutegravir; EGA, estimated gestational age; EVG, elvitegravir; FTC, emtricitabine; ka, absorption rate constant (first-order); M/P, milk to plasma ratio; NVP, nevirapine; PK, pharmacokinetic; PrEP, pre-exposure prophylaxis; PW, pregnant women; Q, intercompartmental clearance; RCT, randomized controlled trial; RFV, efavirenz; TDF, tenofovir disoproxil fumarate; tmax, time to reach maximum (peak) plasma concentration following drug administration; t½, elimination half-life; Vc, central volume of distribution; Vd/F, apparent peripheral volume of distribution; Vp, peripheral volume of distribution; Vss, apparent volume of distribution at steady state; 3TC, lamivudine.
The breastmilk search resulted in 107 citations of which 11 were duplicates. We included four studies in our final analyses (Figure 2). These studies were all conducted in Africa. Three studies included HIV-infected women [36–38] and one included HIV-negative women who received TDF as part of PrEP [17].
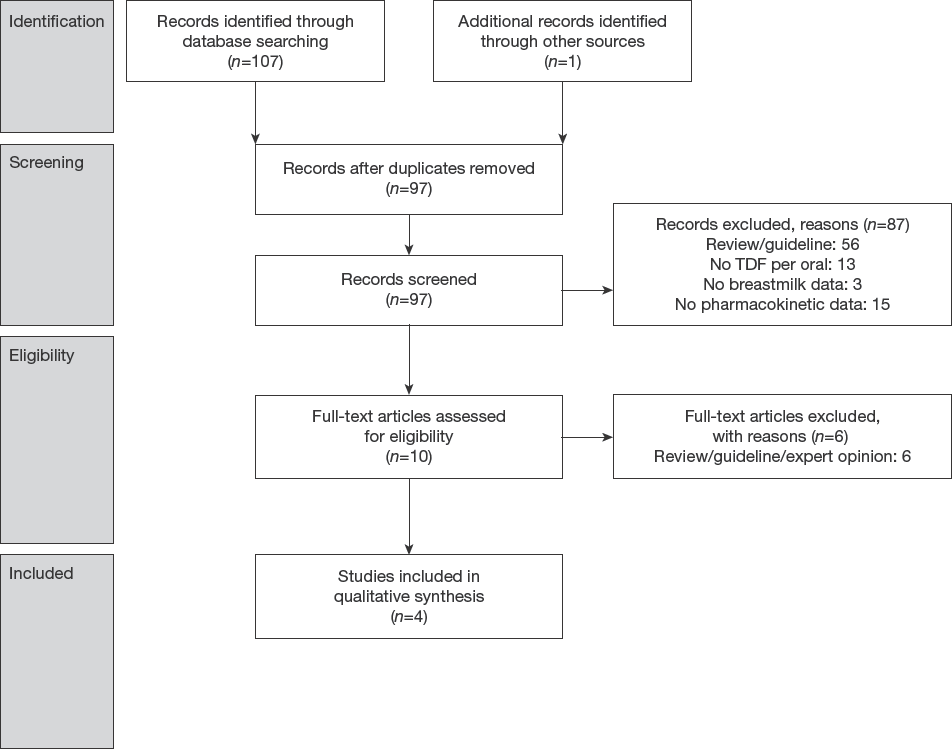
PRISMA flow diagram for PK studies on TDF in breastmilk
Assay methodologies
The PK parameters were tested with high performance liquid chromatography (HPLC) with fluorescence detection which has been validated according to the International Conference of Harmonisation Guidelines in terms of accuracy, precision, specificity, robustness, limits of detection and quantitation, and other aspects of analytical validation [39] or liquid chromatography–mass or tandem-mass spectrometry (LC-MS/MS) methods, validated for the multiplexed quantification of TAF and TFV [40] (Reporting methodology and limit of quantification of tenofovir assay in Additional file 1). The analysed PK samples were of maternal plasma, cord blood, infant plasma, amniotic fluid and breastmilk; and presented as concentration ratios.
Risk of bias within studies
The quality assessment of the studies was based on the GRADE scoring system (in Additional file 1). In this system the studies were scored on study limitations (low, medium or high level), directness, consistency, precision and reporting bias. Most of the studies did not report how patients were selected and did not report the proportion of patients who agreed to participate resulting in medium levels of study limitations. There were no reporting biases detected.
Results of individual studies
Pharmacokinetics during pregnancy
Five publications described steady-state PK parameters [26–30]. A computer-simulated modelling study was also included [25] (Table 2). Of the five steady-state PK studies, four included HIV-infected or non-infected pregnant females treated with 300 mg TDF in steady state, either as PrEP or as treatment, and only Cressey et al. [29] included HBV-infected women. The non-compartmental analyses (NCA) of all publications indicated that the AUC/f and Cmax values were significantly lower in pregnancy compared with non-pregnant women.
Pharmacokinetic outcome measurements of TDF in pregnancy

AUC, area under the plasma concentration–time curve; CI, confidence intervals; Cmax, maximum (peak) plasma drug concentration; C24, plasma drug concentration at time 24; TDF, tenofovir disoproxil fumarate.
The study by Colbers et al. [28] showed that 26% of the patients receiving TDF in pregnancy did not meet the threshold of 2,000 ng/ml*h AUC0–24 (defined as the threshold for TFV efficacy for HIV being the 10th percentile in non-pregnant controls) in the third trimester compared with only 4% of the patients in the post-partum period. One in nine patients that had an AUC below the threshold had a detectable HIV viral load at delivery compared with 6 out of 25 with AUC above the threshold [28].
Colbers et al. [28] showed that the Cmax returned to normal after delivery. Best et al. [27] showed a similar trend, however, this was not statically significantly different between trimesters and the postpartum period. Accordingly, the Clast reported in the various studies was decreased in pregnant women and normalized post-partum [29]. Best et al. and Colbers et al. showed a decrease of 20% and 23% in AUC/f comparing the third trimester and post-partum, respectively [27,28]. These studies estimated an increased oral clearance (Cl/f) of TFV in the third trimester compared with postpartum of 57-55 l/H versus 46-43 l/H, respectively. Thus, the oral clearance (Cl/f) of TFV increased significantly in comparison to post-partum and non-pregnant women and the AUC is inversely correlated with clearance.
The data from these studies [17,18] was used for the computer modelling [25]. The modelling study used population characteristics of a certain patient population and physicochemical parameters of the drug to simulate drug plasma concentrations. Oral clearance (Cl/f) was estimated to increase until an estimated gestational age (EGA) of 28 weeks and then to slowly decrease again. Thus, TFV Cmax, Clast and AUC all decreased in HIV pregnant women in the second and third trimester. The absorption time (Tmax) did not change during pregnancy. Best et al. [27] estimated a terminal t 1/2 of 16.1 h during pregnancy compared with 12.4 h postpartum [27] which suggests that the exposure parameters are affected mainly by an increased volume of distribution, Vd. There were no data on the intracellular phosphorylation kinetics of TFV in pregnancy.
TFV Concentrations
The study from Pyra et al. [30] compared the mean concentrations of TFV during the different trimesters in pregnancy and in pre-pregnant and in non-pregnant women when the drug was provided for PrEP HIV. Their data suggests that after the first trimester the mean concentration of TFV decreases to below the non-pregnant concentration (Figure 3). During pregnancy the TFV concentrations were 45–58% lower compared with non-pregnant after adjusting for adherence. Cressey et al. [29] assessed TFV exposures in HIV-uninfected HBV-infected pregnant women (estimated gestational age 28 weeks), and showed that TFV exposure was 20% lower in pregnancy compared with postpartum. This was similar to the results reported in studies of HIV-infected pregnant women.

TFVmean concentration in pregnant (n=37) and non-pregnant (n=97) women with TDF pre-exposure prophylaxis [30]
Pharmacokinetics during childbirth
Three publications provide PK information after a single oral dose (600 mg or 900 mg) of TDF during childbirth [31–33]. The reported Cmax after 600 mg TDF differed widely among all studies with 234 (range 83–595) ng/ml, 310 (range 70–520) and 448 (range 110–928) ng/ml. The reported AUCs of the women who received 600 mg TDF were 2.73 mg/l*h (range 1.43–3.55) and 4.22 (range 2.77–24.46) mg/l*h. The t 1/2 following the 600 mg dosage was 15.9–19.5 (range 3.3–187.6) h.
Cord blood concentrations
Rimawi et al. [34] compared paired maternal (delivery) and cord blood samples of 10 patients who received a TDF-containing antiretroviral regimen. They found a maternal plasma TDF concentration of 94.6 ng/ml with a cord plasma of 38.5 ng/ml, or a median cord plasma/ maternal plasma ratio of 1:2.5 (95% CI). Yeh et al. [35] measured drug concentrations at delivery in maternal blood plasma, cord blood plasma and amniotic fluid. The TFV concentration in the maternal plasma was 5.0 ng/ml and in cord plasma was 30 ng/ml. These two studies report very different cord plasma/maternal plasma ratios which could be due to the different assays used in the studies, LC-MS/MS and HPLC.
Breast milk
In four studies, maternal plasma and breastmilk samples, taken within 30 min of each other, were compared (Table 3), three studies were done in HIV-infected women. The timing of the samples in relation to the TDF dose varied from 1–2 h after the maternal TDF dose to over 12 h [36–38,41]. The median TDF concentration in breastmilk varied from 3.2 to 14.1 ng/ml with a maternal plasma concentration of 86.7 to 293.0 ng/ml. This resulted in a breastmilk/maternal plasma ratio of 0.03 to 0.07. TFV was excreted in breastmilk in a very low concentration, lower than the concentrations found in cord blood samples, resulting in a median amount of ingested TFV in infants of 0.03% of the recommended infant dose.
Drug concentrations of TDF in breastmilk

Data are median (interquartile range). Cmax, maximum (peak) plasma drug concentration; Cmin, minimum plasma drug concentration; TDF, tenofovir disoproxil fumarate; TFV, tenofovir.
Discussion
In order to prevent MTCT of HBV and HIV in pregnancy, the viral load needs to be low or preferably undetectable, particularly at the time of birth. HBV DNA suppression may be compromised by lower exposure to TDF in later pregnancy. In this review the previously stated threshold for the Cmax of 300 ng/ml for HBV suppression was not achieved. This review shows that the Cmean and Cmax of TFV decrease during pregnancy. With Tmax remaining the same, the absorption of the drug appears to be unaffected. The decrease in Cmax could be a result of the increasing Vd, due to a physiological increase of total body water and extracellular fluid during pregnancy [14]. In one published manuscript this led to a sufficient Cmax in the first trimester, but Cmax then decreased as the pregnancy evolved [14–16]. Drug elimination is increased during pregnancy with increased clearance until a gestation of approximately 28 weeks after which it slowly decreases [27,28,42]. Since TDF is excreted mainly by glomerular filtration, its renal clearance is expected to parallel changes in creatinine clearance during pregnancy [15]. As the AUC is dependent on the clearance, an increase in the creatinine clearance results in a decrease in TFV AUC per trimester of the pregnancy. This review supports conclusions of Benaboud et al. [42] that dosing escalation be considered from 2nd trimester to achieve similar exposure to non-pregnant adults with the caveat that clinical experience of more than 300 mg daily is limited. Dense and sparse PK sampling under TDF treatment correlated with HBV DNA load across gestation and post-partum (or pre-pregnancy) are required to determine if dose adjustment is needed. If this is needed, it needs to be considered that TDF follows a dose-linear PK [9].
Effect of pharmacokinetics on HBV DNA suppression
While this review shows reduced exposure of TDF in pregnancy the implications of this altered PK on HBV DNA suppression are unclear because of unknowns in the PK PD relationships in the initial clearance phase and the treatment duration factor. Previously it has been suggested that the decrease of AUC, Cmax and Cmin that occurs in pregnancy does not affect the viral load in HIV patients [28]. However, the women enrolled were taking multiple antivirals, making it unclear if it was the TDF suppressing the virus or one of the other antivirals. Moreover, co-formulated TDF increases the AUC, Cmax and Cmin of TFV [43].
Treatment with TDF can reduce the HIV or HBV viral load if given for an adequate length of time before delivery. In non-pregnant chronic HBV-infected patients treated with TDF, maximal suppression of HBV DNA plateaus at approximately 32 weeks with good compliance and with a 4-week viral load reduction of approximately 3.9–4.0 log10 IU/ml [44–46]. This reduction is higher than reported in pregnancy, where the HBV DNA decreases between 2.75 and 3.52 log10 IU/ml after 4 weeks of TDF therapy with up to 32% of mothers with an HBV DNA >200,000 IU/ml at delivery [6,47–49]. If started earlier in pregnancy (gestation of 24 weeks), the viral decline can reach 4.08–5.23 log10 IU/ml until childbirth, however, in LIC women tend to present later in pregnancy [49,50]. The HBV DNA viral load needed to omit HBIG is unknown but potentially negated by longer dosing prior to childbirth.
In studies on the PK and PD of TFV in HIV-infected individuals, dose escalating data from 75, 150, 300 and 600 mg showed a dose-proportional increase in viral suppression until 300 mg, the 600 mg dose could not produce a steeper viral decay compared with the 300 mg dosage [44,51]. The Cmax were proportional to the dosage with 375 ng/ml versus 573 ng/ml after 8 h for 300 mg and 600 mg, respectively. The studied plateau TFV concentrations in the healthy individuals were higher compared with the described PK data in pregnancy. This suggests that in healthy individuals the maximum achievable HIV viral decay is with 300 mg but this might be different in pregnancy when the drug concentrations are lower; or different for HBV. Increasing the dosage of TDF to get a Cmax as observed in healthy individuals might produce a steeper viral decay and, as toxicity is related to drug concentrations, would not necessarily increase risks.
More data is needed in the PK/PD of TDF in pregnancy, in particular, on the impact of variable circulating blood concentrations on the clearance rate of HBV. This information could guide recommendations on dosing and the duration of treatment in the prevention of MTCT particularly when optimal delivery of HBIG and birth dose vaccination are compromised.
TDF in Breastfeeding
TFV concentrations in breastfeeding infants are mostly undetectable or lower than what is considered of clinical significance. These findings corroborate with findings from animal studies [52] and suggests that usage of TDF in breastfeeding women is safe for the infant but of no therapeutic value. Most liver disease societies including the American Association for the Study of Liver Diseases and European Association for the Study of the Liver support allowance of breast feeding when mothers were taking TDF. In a resource-constrained setting the risk of infant infection from lactation (cracked nipples) due to HBV rebound (typically 4–8 weeks) after TDF cessation at 1-month post-partum and before protection from infant vaccination is established (when HBIG and birth dose are not accessible), is unknown.
Limitations
This systematic review has several limitations including few available studies most of which are on HIV. The number of studies is disproportionally low compared with the number and ethnicities of pregnant women with HIV and HBV infections worldwide. The two studies reporting on the drug concentrations used different assays to measure the PK which makes comparison difficult. The impact of pharmacogenetics is not reviewed, although it is known that genetic variation of HBV might impact the PK. Lastly, the studies reporting on pharmacokinetics in pregnancy are all based in Europe or USA. This makes the results difficult to translate to patients elsewhere, although no difference in PK has been reported for TFV between these populations in non-pregnant adults.
In conclusion, there are two main highlights of this review: firstly the limited data on the PK of TDF mono-therapy treatment for HBV in pregnant women which suggests lower concentrations of TFV in the second and third trimester of pregnancy compared with the first trimester and non-pregnant status. Secondly, studies to date have occurred in settings where other HBV risk reduction measures (birth dose, HBIG, TDF during lactation) were optimized and as these are not routinely provided in resource-constrained settings consideration for ensuring the most favourable exposure before childbirth is useful.
Footnotes
Acknowledgements
This work was supported by the Wellcome-Trust Major Overseas Program in Southeast Asia (grant number: 106698/Z/14/Z). The funders had no role in the collection, analysis and interpretation of the data, the writing of the article or in submission of the paper for publication. The views expressed in the paper are those of the authors and do not represent the positions of their respective institutions or that of the funding agencies.
EJS declares to have received travels grants from AbbVie and Gilead. The other authors declare no conflicts of interest. The sponsors had no role in the design, execution, interpretation or writing of the study.