
Abstract
Background
Methods
Results
Conclusions
Introduction
HIV continues to be a major global health challenge, infecting more than 36.9 million people worldwide [1]. In 2017, approximately 1.8 million people became infected with HIV, and 0.9 million people died from AIDS-related causes globally [1]. Antiretroviral therapy (ART) has been seminal in reducing the morbidity and mortality associated with HIV type-1 (HIV-1) infection. There are over 25 agents available for use in seven major mechanistic classes of ARTs: nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors, a fusion inhibitor, a C-C chemokine receptor type 5 antagonist, a CD4-directed post-attachment inhibitor, and integrase strand transfer inhibitors [2]. Current guidelines generally recommend three antiviral agents from at least two different mechanistic classes, as treatment with agents across classes has demonstrated sustained virological response [2,3]. Despite the array of therapies currently available, no single antiviral agent or combination of agents is appropriate for every person living with HIV, and there are often additional challenges in finding the most suitable treatment including issues with resistance, tolerability, unfavourable drug–drug interaction (DDI) profiles, high pill burden and/or unfavourable dosing frequency [2,3].
NNRTIs were formerly the cornerstone of front-line therapy; however, as protease inhibitors and integrase strand transfer inhibitors offer a greater barrier to resistance, improved tolerability and more rapid viral suppression for people living with HIV, they are no longer primarily recommended in major international guidelines [2,3]. Although the NNRTIs efavirenz and rilpivirine remain as alternative treatment options under particular clinical circumstances, efavirenz has a relatively high rate of central nervous system-related adverse events (AEs), limiting its tolerability; and rilpivirine has lower virological efficacy, particularly in patients with high baseline HIV-1 RNA (>100,000 copies/ml) and low CD4+ T-cell counts (<200 cells/mm3) [2]. As such, an unmet medical need exists for improved ART, including new NNRTI agents with improved tolerability and efficacy compared with currently available drugs in this class.
Doravirine (DOR, MK-1439) is a novel NNRTI designed to overcome the common resistance mutations which can reduce the effectiveness of other antiretrovirals in this class. Preclinical studies have demonstrated DOR to be active against wild-type HIV-1, as well as the two most prevalent NNRTI-associated mutant viruses (K103N and Y181C substitutions) [4]. In two Phase III studies, DOR demonstrated robust and durable effi-cacy, and was generally well tolerated [5,6]. In the first of these, DOR coadministered with lamivudine (3TC)/tenofovir disoproxil fumarate (TDF) was associated with fewer treatment-emergent central nervous system AEs compared with the combination of efavirenz and emtricitabine/TDF [6]. In the second study, which compared DOR to ritonavir-boosted darunavir when both were coadministered with investigator-selected NRTIs (TDF and emtricitabine or abacavir and 3TC), there were no clinically relevant differences in the incidence of specific AEs, with the exception of a higher incidence of diarrhoea in the darunavir group [5]. In both studies, DOR combination therapy was associated with a more favourable lipid profile and similar antiviral efficacy over 48 weeks of treatment [5,6]. DOR 100 mg administered once daily is indicated for the treatment of HIV-1 infection in combination with other ARTs, including 3TC and TDF, and is available for use as a single tablet or in a fixed dose combination tablet with 3TC and TDF [7,8].
DOR is cleared primarily by oxidative metabolism via cytochrome P450 (CYP)3A [9]. Thus, drugs that induce or inhibit CYP3A may affect DOR elimination; this interaction has been confirmed in clinical DDI studies with the antibiotics rifabutin and rifampin, the anti-fungal ketoconazole, and the antiretrovirals ritonavir and efavirenz [10–14]. DOR was also shown to be a substrate for P-glycoprotein (P-gp) [9]; however, studies conducted to date revealed that P-gp does not have a significant role in DOR absorption or elimination, suggesting that the likelihood of P-gp affecting DOR pharmacokinetics (PK) is minimal [9]. In vitro studies demonstrated that DOR is not expected to have a meaningful impact on the PK of other compounds, including substrates of all major CYPs and drug transporter [15]. Clinical drug-interaction studies with CYP3A and transporter substrates demonstrated no substantive interactions [16–19].
As a commonly used NRTI with a well-characterized PK profile, 3TC is eliminated primarily via urinary excretion by active organic cationic secretion and is not a known perpetrator of DDIs [20]. TDF is another commonly used NRTI which, following absorption, is rapidly converted to its active metabolite, tenofovir, and cleared by renal elimination [21,22]. Although tenofovir has been shown to reduce CYP1A substrate concentrations, it is not a substrate, inducer or inhibitor of CYP3A [21]. Tenofovir DDIs have been reported with didanosine, resulting in increased didanosine concentrations after coadministration [23] and with ritonavir-boosted and unboosted atazanavir, with coadministration resulting in decreased atazanavir plasma concentrations and increased tenofovir concentration [21,24].
Based on the metabolic profiles of DOR, 3TC and TDF, a meaningful PK DDI is unlikely. However, due to the use of these three agents in combination, and the unexpected effects seen with TDF when coadministered with other antiretroviral agents, two clinical trials were conducted to further explore potential DDIs.
Methods
Study design
Study 1 (protocol MK-1439-003) was an open-label, two-period, fixed-sequence study in eight healthy male participants, conducted between 19 September and 23 November 2011. In Period 1, all participants received a single oral dose of DOR 100 mg after an overnight fast. After a washout of ≥7 days, Period 2 began; all participants received a daily dose of TDF 300 mg for 18 days with coadministration of a single dose of DOR 100 mg on day 14. All doses of TDF alone were administered within 30 min prior to or after a standard meal; on day 14, study drugs were coadministered in the fasted state.
Study 2 (protocol MK-1439-038) was an open-label, single-dose, randomized, three-period crossover study in 15 healthy participants, conducted in January 2015. In the three treatment periods, participants received the following in a randomized manner: a single oral dose of DOR 100 mg; coadministration of single oral doses of 3TC 300 mg and TDF 300 mg; and coadministration of single oral doses of DOR 100 mg, 3TC 300 mg and TDF 300 mg. Study drugs were administered after an overnight fast. The washout period between drug administrations was ≥7 days.
The studies were conducted in accordance with principles of Good Clinical Practice and were approved by the appropriate institutional review boards (Study 1: Thomas Jefferson University IRB, Philadelphia, PA, USA; Study 2: the Institutional Review Board of Optimum Clinical Research Inc., Oshawa, ON, Canada) and regulatory agencies.
Study populations
Study 1 included healthy men, 18–50 years of age with a body mass index ≤35 kg/m2. Study 2 included healthy men and women, 18–65 years of age with a body mass index of 19–33 kg/m2. In both studies, participants with a history of clinically significant medical conditions, estimated creatinine clearance of ≤80 ml/min (based on Cockcroft–Gault equation), drug or alcohol abuse, recent smoking or positive test for HIV, or who were hepatitis B or C positive, were excluded. Concomitant medications were not permitted from 14 days or 5 half-lives prior to the start of the trials until trial completion (although participants could receive concomitant therapy and continue in the study if the sponsor and investigator agreed). Participants in both studies provided written, informed consent prior to any study-related procedures being performed.
Sample collection and plasma concentration determination
In Study 1, blood samples for assay of DOR plasma concentration were obtained pre-dose and up to 120 h following administration of DOR on day 1 (Period 1), and coadministration of DOR and TDF on day 14 (Period 2). In Study 2, blood samples were collected pre-dose and up to 72 h post-dose.
In both studies, DOR plasma concentrations were analysed by liquid–liquid extraction for analyte isolation followed by liquid chromatographic–tandem mass spectrometric (LC-MS/MS) detection using a validated method (MSD, Oss, the Netherlands) [13]. The lower limit of quantitation was 1 ng/ml. The analytical range of the assay was 1.00–1,000 ng/ml. For Study 1, the inter-day accuracy of the quality control samples was 103.3–105.0%, and the inter-day precision was 3.3–5.2%. For Study 2, the inter-day accuracy was 97.0–99.5%, and the inter-day precision was 3.5–5.3%. In Study 2, following extraction, the plasma concentrations of 3TC and tenofovir were determined by validated achiral LC-MS/MS detection methods (Pharma Medica Research, Inc., Mississauga, ON, Canada). The analytical ranges of the assays were 5.00–3,000 ng/ml for 3TC and 2.00–500 ng/ml for tenofovir. For 3TC, the inter-day accuracy of the quality control samples was 97.8–105.2%, and the inter-day precision was 0.9–3.3%. For tenofovir, the inter-day accuracy was 98.5–101.5% and the inter-day precision was 1.0–2.2%.
PK Evaluations
In Study 1, DOR area under the concentration–time curve from time 0 extrapolated to infinity (AUC0–∞), maximum plasma concentration (Cmax), time to reach maximum plasma concentration (Tmax) and the apparent terminal half-life (t1/2; calculated as the quotient of the natural log of 2 [ln (2)] and apparent terminal elimination rate constant) were calculated using Phoenix® Win-Nonlin® (Version 6.3; Certara, Princeton, NJ, USA). The observed plasma concentrations at 24 h post-dose (C24 h) were obtained directly from plasma concentrations using SAS (Version 9.3; SAS Institute Inc., Cary, NC, USA). In Study 2, values of the same PK parameters as in Study 1 were calculated for DOR, 3TC and tenofovir using the non-compartmental approach in Phoenix® WinNonlin®.
Safety and tolerability
Safety and tolerability were assessed in both studies by physical examinations, vital signs, laboratory assessments and AE monitoring.
Statistics
In both studies, the individual values of AUC0–∞, Cmax and C24 h were ln-transformed prior to analysis and evaluated separately using a linear mixed-effect model. In Study 1, treatment was a fixed effect and subject was a random effect. A two-sided 90% CI for the geometric mean ratio (GMR; DOR+TDF/DOR alone) was generated for DOR AUC0–∞, Cmax and C24 h from the mixed-effect model. Tenofovir PK were not analysed. Descriptive statistics were provided for Tmax and apparent t1/2. Median values were reported for Tmax while the geometric mean was reported for t1/2.
In Study 2, AUC0–∞, Cmax and C24 h were analysed using a linear mixed-effect model appropriate for a three-period, two-treatment crossover design with fixed-effect terms for treatment and period. An unstructured covariance matrix was used to allow for unequal treatment variances and to model the correlation between different treatment measurements within the same subject via the REPEATED statement SAS PROC MIXED. Kenward and Roger's method was used to calculate the denominator degrees of freedom for the fixed effects (DDFM=KR).
A two-sided 90% CI for the GMRs (DOR+3TC+ TDF/DOR alone) was generated for DOR AUC0–∞, Cmax and C24 h.
In addition, 95% CIs were generated from the above mixed-effect model for geometric means by treatment for DOR AUC0–∞, Cmax and C24 h. 3TC and tenofovir AUC0–∞, Cmax and C24 h after coadministration of DOR 100 mg, 3TC 300 mg and TDF 300 mg were analysed in a similar manner.
Results
Study populations
A total of eight healthy male participants were enrolled in Study 1; one participant discontinued on day 11 in Period 2 due to an AE that was not study-drug related. A total of 15 participants were enrolled in Study 2, all of whom completed the study. Demographics for participants from both studies are summarized in Table 1.
Study population demographics

PK Evaluations
Study 1: mean plasma concentration–time curves for DOR alone or coadministered after multiple doses of TDF are shown in Figure 1A. DOR PK summary statistics are listed in Table 2. The GMRs (90% CI) of DOR AUC0–∞ and C24 h (DOR+TDF/DOR alone) were 0.95 (0.80, 1.12) and 0.94 (0.78, 1.12), respectively. The GMR (90% CI) of DOR Cmax was 0.80 (0.64, 1.01). Tmax and apparent t1/2 were similar between the two treatment groups.

Plasma concentration–time profiles
Plasma PK of DOR 100 mg administered alone or with multiple-dose TDF 300 mg administered once daily for 14 days to healthy participants
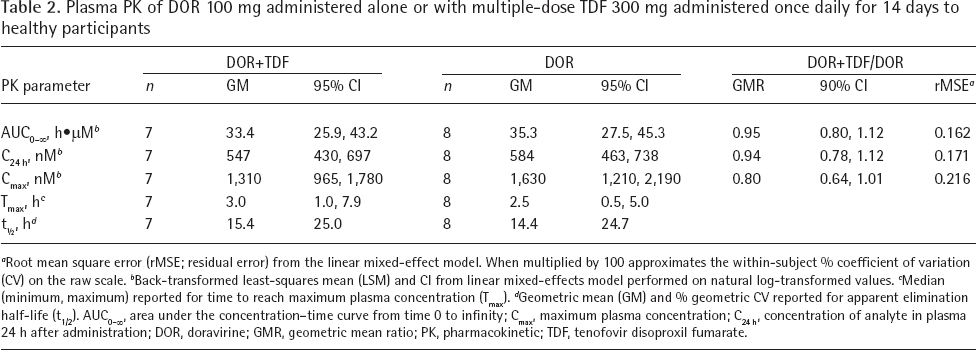
Root mean square error (rMSE; residual error) from the linear mixed-effect model. When multiplied by 100 approximates the within-subject % coefficient of variation (CV) on the raw scale.
Back-transformed least-squares mean (LSM) and CI from linear mixed-effects model performed on natural log-transformed values.
Median (minimum, maximum) reported for time to reach maximum plasma concentration (Tmax).
Geometric mean (GM) and % geometric CV reported for apparent elimination half-life (t1/2). AUC0–∞, area under the concentration–time curve from time 0 to infinity; Cmax, maximum plasma concentration; C24 h, concentration of analyte in plasma 24 h after administration; DOR, doravirine; GMR, geometric mean ratio; PK, pharmacokinetic; TDF, tenofovir disoproxil fumarate.
Study 2: the mean plasma concentration–time pro-files for DOR, 3TC and tenofovir following DOR or 3TC+TDF administration or DOR+3TC+TDF coadministration are shown in Figure 1B–1D. DOR, 3TC and tenofovir PK summary statistics are listed in Tables 3 and 4. The GMRs (90% CI) of DOR AUC0–∞ and Cmax (DOR+3TC+TDF/DOR alone) were 0.96 (0.87, 1.06) and 0.97 (0.88, 1.07), respectively. The GMRs (90% CI) of 3TC AUC0–∞ and Cmax (DOR+3TC+TDF/3TC+TDF) were 0.94 (0.88, 1.00) and 0.92 (0.81, 1.05), respectively. GMRs (90% CI) of tenofovir AUC0–∞ and Cmax (DOR+3TC+TDF/3TC+TDF) were 1.11 (0.97, 1.28) and 1.17 (0.96, 1.42), respectively. Individual PK ratios and corresponding GMR plots of DOR, 3TC and tenofovir with and without coadministration of companion agents are shown in Figure 2.

Individual plasma pharmacokinetic ratios
Plasma PK of DOR 100 mg administered alone or with single-dose TDF 300 mg and 3TC 300 mg to healthy participants

Estimated based on the elements of the variance-covariance matrix as: coefficient of variation (CV; %) = 100*sqrt[(σA 2 + σB 2 - 2*σAB)/2].
Back-transformed least-squares mean (LSM) and CI from linear mixed-effects model performed on natural log-transformed values.
Median (minimum, maximum) reported for time to reach maximum plasma concentration (Tmax).
Geometric mean (GM) and % geometric CV reported for apparent elimination half-life (t1/2). AUC0–∞, area under the concentration–time curve from time 0 to infinity; Cmax, maximum plasma concentration; C24 h, concentration of analyte in plasma 24 h after administration; DOR, doravirine; GMR, geometric mean ratio; PK, pharmacokinetic; TDF, tenofovir disoproxil fumarate; 3TC, lamivudine.
Plasma PK of 3TC and tenofovir following single-dose administration of 3TC 300 mg + TDF 300 mg or DOR 100 mg + 3TC 300 mg + TDF 300 mg to healthy participants
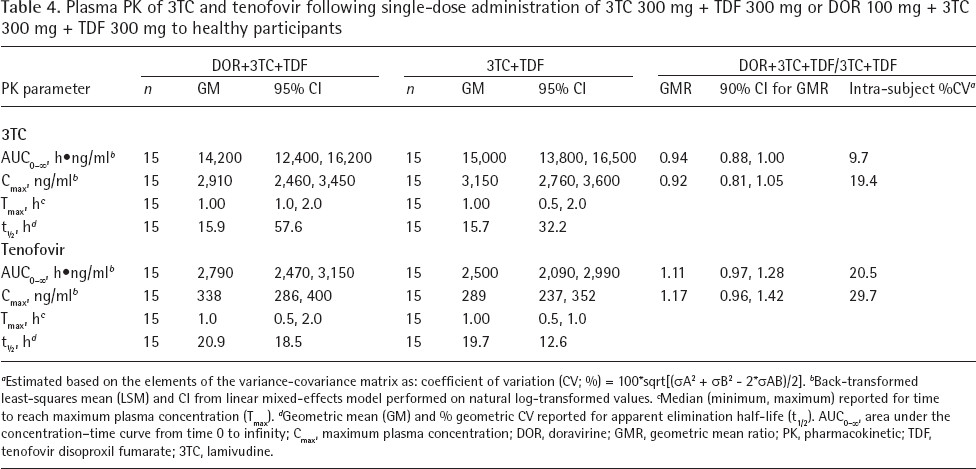
Estimated based on the elements of the variance-covariance matrix as: coefficient of variation (CV; %) = 100*sqrt[(σA 2 + σB 2 - 2*σAB)/2].
Back-transformed least-squares mean (LSM) and CI from linear mixed-effects model performed on natural log-transformed values.
Median (minimum, maximum) reported for time to reach maximum plasma concentration (Tmax).
Geometric mean (GM) and % geometric CV reported for apparent elimination half-life (t1/2). AUC0–∞, area under the concentration–time curve from time 0 to infinity; Cmax, maximum plasma concentration; DOR, doravirine; GMR, geometric mean ratio; PK, pharmacokinetic; TDF, tenofovir disoproxil fumarate; 3TC, lamivudine.
Safety
All treatment combinations were generally well tolerated. There were no serious AEs, events of clinical interest or deaths reported during the studies. All AEs were mild in intensity, of limited duration and resolved by the end of the study.
In Study 1, three of the eight participants (37.5%) reported a total of ten AEs, four of which were considered to be related to study treatment (fatigue, [DOR alone], headache [DOR alone], rash [TDF] and somnolence [DOR+TDF]). Headache was the only AE reported by more than one participant. One participant was discontinued on day 11 of Period 2 due to an AE that was not study-drug related.
In Study 2, 5 of the 15 participants (33.3%) reported a total of 5 AEs. One incidence of somnolence (DOR alone) and one of headache (DOR+3TC+TDF) were considered to be related to study treatment.
Discussion
There is a continuing need for improved therapeutics for the treatment of HIV-1 infection. DOR is a novel HIV-1 NNRTI that is indicated for use in combination with other antiretroviral agents, and as a fixed-dose regimen with 3TC and TDF as a complete regimen, for the treatment of HIV-1 infection in adults with no prior antiretroviral treatment history [7,8]. Although the metabolic profiles of these agents do not suggest that there would be meaningful DDIs with coadministration, clinical investigation was pursued to further evaluate potential DDIs.
Data from the two studies reported here demonstrate that neither coadministration of multiple doses of TDF nor coadministration with single doses of 3TC+TDF (at the recommended therapeutic dose of 300 mg each for 3TC and TDF) have a clinically meaningful impact on DOR PK. This is evidenced by a lack of a meaningful effect on DOR AUC0–∞, Cmax and C24 h, with AUC0–∞ and C24 h GMRs close to unity and Cmax reduced by 20% following multiple doses of TDF. The minor reduction in DOR Cmax is not anticipated to have any meaningful impact on DOR efficacy or safety, as a DOR Phase IIb trial demonstrated similar efficacy to efavirenz across a range of doses from 25 to 200 mg [25]. The single-dose DDI assessment in Study 2 further supports a lack of interaction, with DOR AUC0–∞, Cmax and C24 h all without clinically meaningful changes.
3TC and TDF are commonly administered together, without evidence of a meaningful interaction when coadministered [20,21]. As such, Study 2 was designed with coadministration of 3TC+TDF without evaluation of each of the separate components. Data showed the lack of a meaningful effect of DOR on either 3TC or tenofovir PK. Tenofovir exposure and Cmax increased slightly (by 11% and 17%, respectively) with coadministration of DOR. These changes are not clinically meaningful, based on drug-interaction effects and dosing recommendations for TDF [21]. The cause of the effect is unknown. It has been noted that tenofovir is a substrate of P-gp and breast cancer-resistant protein (BCRP) transporters [21]. Atazanavir and other HIV protease inhibitors, which are P-gp and BCRP inhibitors, modestly increase tenofovir plasma concentrations, although not to a clinically meaningful level, likely secondary to P-gp and BCRP inhibition [21,26,27]. However, in vitro observations with DOR have shown that it is not an inhibitor of P-gp, indicating that interactions between DOR and P-gp are unlikely to be the cause of the increases to plasma tenofovir levels in the current study [15].
Study 1 was designed to assess the impact of TDF at steady state on DOR PK to maximize any potential inductive or time-dependent effects of TDF. While Study 2 was conducted with single-dose administration only, no inductive effects by DOR, 3TC or tenofovir were anticipated and there is no time dependence for the PK of DOR, 3TC or tenofovir. Moreover, with Study 1 data demonstrating minimal effect of TDF on DOR PK, a single-dose assessment was considered an appropriate approach for Study 2 and is anticipated to be predictive of multiple-dose behaviour [28]. The results of these studies did not demonstrate any substantive effect and indicate that, with multiple-dose administration, there would not be a meaningful PK DDI between these agents.
Administration of DOR, 3TC and TDF individually and in combination was generally well tolerated, providing further evidence of the tolerability of DOR alone and in combination with 3TC and TDF. The most common treatment-related AEs in both Study 1 and Study 2 were headache and somnolence, which have also been reported in DOR Phase III studies [5,6]. The lack of DDIs between DOR and 3TC+TDF supports the fixed-dose, three-drug, single-tablet regimen (MK-1439A [DOR 100 mg/3TC 300 mg/TDF 300 mg]) that has been developed [29], and which was the formulation used in a recently reported Phase III study (discussed in the Introduction) [6].
In summary, multiple doses of TDF coadministered with a single dose of DOR did not have a clinically meaningful effect on the PK of DOR. DOR, 3TC and tenofovir PK were similar when administered alone or coadministered. Consequently, coadministration of the three drugs without dose adjustment is supported.
Footnotes
Acknowledgements
The authors would like to thank the trial staff and participants. In addition, the authors would like to thank Paul Fackler and Marty Behm (both formerly Merck & Co., Inc., Kenilworth, NJ, USA) for their help with the study. Medical writing and editorial assistance, under the direction of the authors, was provided by Annette Smith of CMC AFFINITY, a division of McCann Health Medical Communications Ltd., Macclesfield, UK, in accordance with Good Publication Practice (GPP3) guidelines. This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
This research was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. JG, LF, KLY, IT and MI are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and may own stock and/or hold stock options in Merck & Co., Inc., Kenilworth, NJ, USA. MSA, CR, YG and RL were employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA at the time the study was conducted. WKK has no conflicts of interest to disclose.