
Abstract
Background
Methods
Results
Conclusions
Introduction
Currently, each of the four non-nucleoside reverse transcriptase inhibitors (NNRTIs) for the treatment of HIV-1 infection has limitations. Efavirenz is associated with frequent central nervous system (CNS) adverse events (AEs) [1], including increased risk of suicidality [2]. Rilpivirine must be taken with food, has significant interactions with acid-reducing agents, and has limited efficacy in individuals with high baseline HIV-1 RNA (>100,000 copies/ml) and low CD4+ T-cell counts (<200 cells/mm3) [3–5]. Etravirine is not approved for first-line treatment and requires twice-daily dosing [6]. Finally, nevirapine is associated with skin and liver hypersensitivity reactions, and has prescribing restrictions based on gender and CD4+ T-cell counts [7]. These limitations have led to changes in recommendations for NNRTI-containing regimens from preferred to alternative first-line antiretroviral therapy (ART) in most international guidelines [3,4,8,9]. Alternative NNRTI treatment options with both high efficacy and tolerability are needed.
Doravirine (MK-1439) is an NNRTI that has high in vitro potency versus a broad panel of isolates, including some common NNRTI-resistant variants [10]. In Phase I studies, doravirine demonstrated good tolerability across a wide dose range and a pharmacokinetic pro-file suitable for once-daily dosing, irrespective of food intake and coadministration of antacids or acid-reducing agents [11–13]. Doravirine is a substrate for cytochrome P450 (CYP450) 3A-mediated metabolism [14]; coadministration with strong CYP450 3A inducers may significantly decrease doravirine plasma levels [15,16]. Drug–drug interactions via other major drug-metabolizing enzymes and transporters are unlikely [14], enabling coadministration with a wide range of treatments for HIV-1 infection and associated concurrent illnesses [11,14]. In ART-naive adults with HIV-1 infection, doravirine monotherapy given for 7 days demonstrated robust antiviral activity, without evidence of viral resistance [17]. Here we present the final results of a Phase IIb, dose selection trial that compared the safety and efficacy of doravirine with that of efavirenz (both administered with tenofovir disoproxil fumarate [TDF]/emtricitabine [FTC]) in ART-naive adults with HIV-1 infection. The 24-week and 48-week interim results of this trial have been previously reported [18–21].
Methods
Study design
This Phase IIb, randomized, double-blind trial (MK-1439 Protocol 007; NCT01632345) consisted of two parts: a 24-week dose selection phase (Part I), followed by comparison of the safety and efficacy of the selected dose of doravirine versus efavirenz for 96 weeks (Part II; Additional file 1). Participants were enrolled at 63 centres across Australia, Belgium, Canada, France, Germany, the Netherlands, Puerto Rico, Romania, Russia, Spain and the US. The trial was conducted in accordance with principles of Good Clinical Practice and was approved by the appropriate institutional review boards and regulatory agencies. All participants provided written informed consent.
Participants
Eligible participants were adults (≥18 years of age) with plasma HIV-1 RNA ≥1,000 copies/ml, CD4+ T-cell count ≥100 cells/mm3 and no prior ART experience. Participants were required to be clinically stable for ≥2 weeks before the trial, with no signs or symptoms of acute infection (non-HIV-1) and no significant laboratory abnormalities within 45 days of treatment. Those with known HIV-1 genotypic resistance to FTC, TDF or efavirenz, or active hepatitis B or C infection were excluded. Additional exclusion criteria are shown in Additional file 2.
Randomization and assessment
Participants were stratified by screening HIV-1 RNA ≤100,000 or >100,000 copies/ml prior to randomization. In Part I, participants were randomized 1:1:1:1:1 to receive doravirine (Merck & Co., Inc., Kenilworth, NJ, USA) 25 mg, 50 mg, 100 mg or 200 mg once daily; or efavirenz 600 mg once daily, both in combination with co-formulated TDF 300 mg/FTC 200 mg (Gilead Sciences Inc., Foster City, CA, USA), for up to 96 weeks. Following the interim analysis for dose selection at week 24 in Part I, additional participants were randomized 1:1 in Part II to receive the selected dose of doravirine (100 mg) or efavirenz 600 mg once daily, each in combination with TDF/FTC, for 96 weeks. Participants receiving the non-selected doses of doravirine in Part I were switched to the selected dose at the next planned study visit. For both parts of the trial, efavirenz and open-label TDF/FTC were administered according to the prescribing information [1,22].
A combination of active drugs and placebo was given to maintain blinding of doravirine and efavirenz: participants took six tablets per day before dose selection (four doravirine or matching placebo, one efavirenz or matching placebo, one TDF/FTC) and three tablets per day after dose selection. Doravirine (or placebo) was to be taken in the morning. Open-label TDF/FTC was to be taken in the morning with food. Efavirenz (or placebo) was to be taken at bedtime without food on an empty stomach.
Participants visited the study site at days 1 and 14; weeks 4, 8, 12, 16 and 24; and every 12 weeks thereafter up to week 96. At each visit, participants were assessed through physical examination, laboratory tests including haematological analysis, serum chemistry tests, fasting lipid parameters, CD4+ T-cell counts and measurement of HIV-1 RNA (Abbott RealTime HIV-1 assay; Abbott Laboratories, IL, USA; lower limit of quantification: 40 copies/ml). Clinical AEs were assessed at every visit by patient report. No questionnaires were used to solicit specific symptoms, including for CNS events. Medication compliance was monitored with diary cards completed by the participants and returned at each study visit.
Protocol-defined virological failure (PDVF) included two categories: non-response (participants who did not achieve HIV-1 RNA <40 copies/ml by week 24) and rebound (initial response of HIV-1 RNA <40 copies/ml, followed by two consecutive measurements of HIV-1 RNA ≥40 copies/ml at least 1 week apart at or after week 24). Participants with PDVF and sufficient viral RNA (>400 copies/ml) in the confirmatory plasma sample were assessed for genotypic and phenotypic viral resistance (Monogram Biosciences, South San Francisco, CA, USA). Participants with PDVF were recommended, but not required, to discontinue the study.
Outcomes
Efficacy and safety outcomes were assessed for each doravirine dose versus efavirenz in Part I, and for the selected dose of doravirine (100 mg) versus efavirenz using data from these groups in Part I and Part II combined (hereafter referred to as Parts I+II). Since there was no clear evidence of heterogeneity across the doravirine dose groups, data displays include an additional doravirine group that includes all participants randomized to any dose of doravirine in Part I.
The primary efficacy outcome was the proportion of participants with HIV-1 RNA <40 copies/ml at week 24 in Part I and in Parts I+II. Secondary efficacy outcomes were the proportion of participants with HIV-1 RNA <200 copies/ml and the change from baseline in CD4+ T-cell count.
Safety was assessed by physical examinations, laboratory tests, 12-lead electrocardiogram, and recording of AEs coded using the Medical Dictionary for Regulatory Activities (MedDRA; version 17.1). Changes in laboratory values were classified according to the Division of AIDS criteria [23]. The primary safety end point was the proportion of participants experiencing CNS events (from the following pre-specified list of MedDRA terms) by weeks 8 and 24 of Parts I+II: dizziness, sleep disorders (abnormal dreams, insomnia, nightmare), altered sensorium (concentration impaired, confusional state, delirium, depressed level of consciousness, disturbance in attention, somnolence), depression/suicide (depressed mood, depressive symptom, depression, major depression, suicidal behaviour, suicidal ideation, suicide attempt, completed suicide), psychotic disorders (acute psychosis, hallucination [including auditory and/or visual], psychotic disorder), or nervous system disorder. All such events regardless of attribution were pooled and evaluated as CNS events.
Statistical analysis
The primary objective of the study was to support the doravirine dose selection for the Phase III clinical programme. The primary population for the efficacy analyses included all randomized participants who had at least one post-randomization observation after receiving at least one dose of blinded study treatment. The non-completer = failure (NC=F) approach to missing data was used for the primary and secondary virological response end points (participant proportions with HIV-1 RNA <40 copies/ml and <200 copies/ml, respectively). All missing values were considered failures with the exception of intermittent missing values flanked by two successes, which were excluded. Sensitivity analyses were conducted for the primary end point using the following missing data approaches: treatment-related discontinuation = failure (discontinuations due to lack of efficacy or AEs were considered failures thereafter), observed failure (OF; discontinuations due to lack of efficacy were considered failures thereafter), and the FDA snapshot method [24].
For all virological end points in Part I and Parts I+II, the difference in proportion of participants with virological responses between treatment groups and the associated 95% CIs were calculated using Miettinen and Nurminen's method [25] with stratification by screening HIV-1 RNA ≤100,000 or >100,000 copies/ml. To assess the consistency of the treatment effect in Parts I+II, the between-group differences for the primary end point were estimated within subgroups defined by baseline HIV-1 RNA level (≤ or >100,000 copies/ml) using the OF approach.
For the secondary end point of change from baseline in CD4+ T-cell counts, descriptive statistics including 95% CIs were provided and missing data were handled using the OF approach. Baseline values were carried forward for participants who discontinued due to lack of efficacy.
The safety analyses used the ‘all patients as treated’ population, including all randomized participants who received at least one dose of study treatment. The primary hypothesis of Parts I+II was that doravirine at the final selected dose is superior to efavirenz, each in combination with TDF/FTC, as measured by proportion of participants with CNS events by week 8 (with hypothesis testing also at week 24 if superior at week 8). CNS events occurring by week 8 and week 24 in Parts I+II were subjected to inferential testing for statistical significance with P-values and 95% CIs provided for between-group comparisons.
Sample size was calculated according to the power for comparing the pre-specified CNS AEs between doravirine at the selected dose and efavirenz; this determined the size of cohort II. With 100 participants per treatment group in Parts I+II, and assuming the CNS event rates were 20% for doravirine and 52% for efavirenz by week 8, the trial had >99% power, with 95% confidence, to detect whether the selected dose of doravirine had a significantly lower rate of CNS events than efavirenz. Assuming CNS event rates of 30% for doravirine and 50% for efavirenz, 100 participants per arm provided 83% power, with 95% confidence, to demonstrate superiority of doravirine over efavirenz.
Results
The trial was conducted between 15 October 2012 and 21 March 2016. Participant disposition for both parts are shown in Additional file 3. Of the 462 individuals screened, 342 were enrolled and randomized across 63 sites. Demographics and baseline clinical characteristics were generally well balanced across treatment groups (Table 1). In Part I, 26.9% of doravirine participants (range 19.5% [200 mg] to 34.9% [50 mg]) and 30.2% of efavirenz participants discontinued study therapy before week 96 (Additional file 3). In Part II, 16.7% of participants in each treatment group discontinued study therapy before week 96 (Additional file 3).
Baseline demographics and participant characteristics (Parts I+II)

Note: both doravirine and efavirenz were administered with tenofovir disoproxil fumarate/emtricitabine.
Other race includes: American Indian or Alaska Native, multiracial and unknown (missing data).
The proportion of participants with HIV-1 RNA <40 copies/ml at week 24 of Part I was over 70% for all doravirine doses (25 mg, 80.0% [95% CI, 64.4,90.9]; 50 mg, 74.4% [58.8,86.5]; 100 mg, 71.4% [55.4,84.3]; 200 mg, 80.5% [65.1,91.2]) compared with 64.3% (48.0, 78.4) for efavirenz, with similar results observed regardless of missing data approach (Additional file 4). The proportion of participants with HIV-1 RNA <200 copies/ml at week 24 ranged from 83.7–92.9% in the doravirine groups compared with 81.0% for efavirenz. The mean change from baseline in CD4+ T-cell count at week 24 of Part I ranged from 113–154 cells/mm3 in the doravirine groups compared with 121 cells/mm3 for efavirenz.
Doravirine was generally well tolerated at all doses studied in Part I (Table 2). There was no apparent pattern of dose-related toxicity. The proportion of participants with drug-related AEs was lower in each of the doravirine groups (16.7–46.5%) versus the efavirenz group (57.1%). In total, 3.0% of participants receiving any dose of doravirine discontinued due to AEs compared with 4.8% of those receiving efavirenz. Through week 24 of Part I, the most common laboratory abnormalities were primarily Grade 1 changes (Additional file 5).
Summary of clinical AEs up to week 24 (Part I)
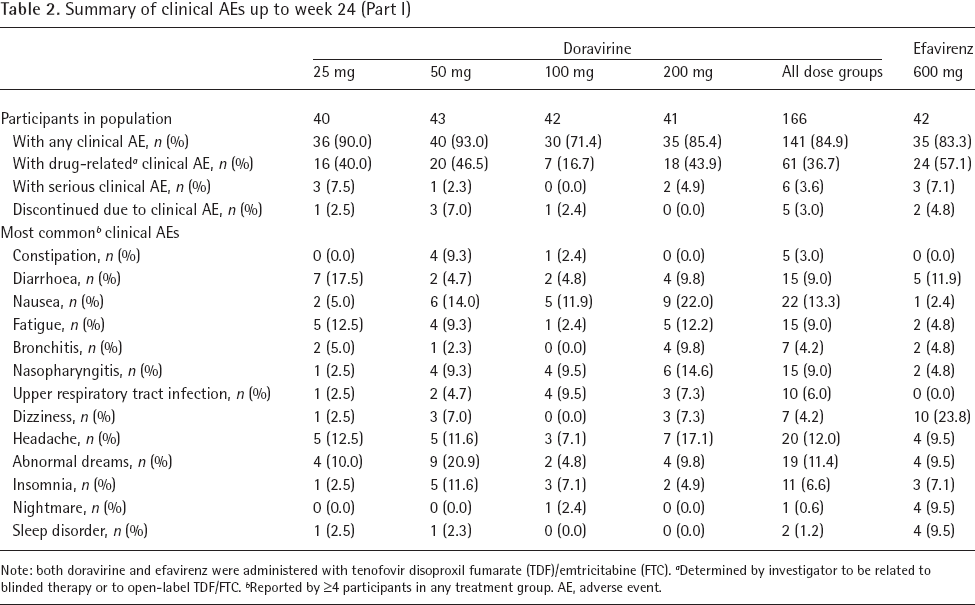
Note: both doravirine and efavirenz were administered with tenofovir disoproxil fumarate (TDF)/emtricitabine (FTC).
Determined by investigator to be related to blinded therapy or to open-label TDF/FTC.
Reported by ≥4 participants in any treatment group. AE, adverse event.
Based on Part I week 24 efficacy and safety results, doravirine 100 mg was selected for enrolment in Part II. Participants who received doravirine at 25, 50 and 200 mg doses were all switched to doravirine 100 mg after dose selection; however, the data shown below for doravirine 100 mg from Parts I and II combined includes only those Part I participants randomized to 100 mg, plus all Part II participants randomized to doravirine, all of whom received the 100 mg dose. In Parts I+II, the proportion of participants with HIV-1 RNA <40 copies/ml at week 24 was 72.9% for doravirine 100 mg versus 73.1% for efavirenz (difference [95% CI], −0.5 [-12.3,11.2]). Similar response rates were observed at subsequent time points through week 96 (Figure 1A) and were similar between treatment groups regardless of missing data approach (Additional file 6). The proportion of participants with HIV-1 RNA <200 copies/ml was 89.7% and 79.6% at weeks 24 and 96, respectively, for doravirine 100 mg compared with 87.0% and 75.9% at weeks 24 and 96, respectively, for efavirenz. The mean change in CD4+ T-cell counts increased throughout the study in both treatment groups, reaching 259 cells/mm3 for doravirine 100 mg and 264 cells/mm3 for efavirenz at week 96 (Figure 1B).

Primary and secondary end points over time, up to week 96, Parts I+II
In the subgroup analyses of Parts I+II, the proportion of participants with baseline HIV-1 RNA ≤100,000 copies/ml who achieved HIV-1 RNA <40 copies/ml at week 96 was similar between doravirine 100 mg and efavirenz (88.9% [95% CI 78.4,95.4] versus 87.3% [76.5,94.4], treatment difference 1.6 [-10.4,13.7]). Among participants with baseline HIV-1 RNA >100,000 copies/ml, the proportion who achieved HIV-1 RNA <40 copies/ml at week 96 was 78.1% (60.0, 90.7) for doravirine 100 mg versus 93.1% (77.2, 99.2) for efavirenz (difference −15.0 [-33.4,3.5]).
Over the course of the trial, 43 doravirine recipients (26 non-responders [11.2%] and 17 rebounders [7.3%]) and 14 efavirenz recipients (10 non-responders [9.3%] and 4 rebounders [3.7%]) met the criteria for PDVF. In the doravirine 100 mg group, 19 (17.6%) participants met criteria for PDVF, including 16 (14.8%) non-responders. A substantial proportion of the non-responders achieved virological suppression later in the trial: 12/26 of the doravirine recipients and 5/10 of the efavirenz recipients at week 48; 16/26 and 8/10, respectively, at week 96.
In total, 18 participants with PDVF had samples with sufficient HIV-1 RNA (>400 copies/ml) for resistance testing. Two participants in the doravirine 25 mg group showed genotypic resistance associated with NNRTIs (K101K/E, P236P/L mutations in one participant, and V106V/I, F227C mutations in one participant), and one participant in the 100 mg group showed mutations associated with resistance to NNRTIs (E138E/G, V179D) and nucleoside reverse transcriptase inhibitors (A62V). Only the virus with V106V/I, F227C mutations displayed phenotypic resistance to doravirine, with a 66-fold decrease in susceptibility; this participant also developed resistance to FTC (M184V). Two of the three efavirenz participants with PDVF had virus with efavirenz resistance mutations (K101K/Q, G190S in one participant, and K103N, N348I in one participant) and phenotypic resistance to efavirenz.
The occurrence of pre-specified CNS events by week 8 and 24 in Parts I+II was a key safety end point of the trial. At week 8, the proportion of participants with CNS events was significantly lower in the doravirine 100 mg group (24.1%) compared with the efavirenz group (44.4%; P=0.002; difference −20.4%; 95% CI −32.4, −7.8). The difference was also observed through week 24, with 26.9% and 47.2% of participants who received doravirine 100 mg and efavirenz, respectively, reporting at least one of the pre-specified CNS events (P=0.002; difference −20.4%; 95% CI −32.6, −7.5). At both time points, dizziness and abnormal dreams accounted for most of the difference between doravirine 100 mg and efavirenz (Figure 2).

CNS events in Parts I+II at week 8 and week 24
Through week 96 of Parts I+II, one or more AEs occurred in 89.8% of the doravirine 100 mg group and 96.3% of the efavirenz group and led to discontinuation in 4.6% and 10.2%, respectively (Table 3). No deaths occurred during the study. The proportion of participants with drug-related AEs in Parts I+II was lower for doravirine 100 mg (35.2%) versus efavirenz (58.3%; difference [95% CI] −23.1 [-35.6,-9.9]). Overall, the most commonly reported drug-related AEs were dizziness (7.4% versus 26.9%), abnormal dreams (5.6% versus 14.8%) and nausea (7.4% versus 6.5%) for doravirine 100 mg and efavirenz, respectively. The proportion of participants with treatment-emergent laboratory abnormalities of Grade 2 or higher was similar between doravirine 100 mg and efavirenz through week 96 of Parts I+II, with two exceptions: Grade 2 elevations in total cholesterol and low-density lipoprotein (LDL)-cholesterol were less common with doravirine 100 mg (0.0% and 2.0%, respectively) than with efavirenz (9.7% and 9.8%, respectively; Additional file 7). For the overall doravirine group (all dose groups combined), Grade 2 elevations in total cholesterol and LDL-cholesterol were also less common (0.9% and 1.4%, respectively) than in the efavirenz group. Grade 3 total cholesterol and Grade 3 LDL-cholesterol events occurred in 2 participants in the efavirenz group only (no Grade 4 events). Grade 2 creatinine elevations were more common in the overall doravirine group (8.3%) than in the efavirenz group (0.9%). No Grade 3 or 4 creatinine elevations and no discontinuations due to renal AEs occurred in any treatment group.
Summary of clinical AEs up to week 96 (Parts I+II)

Note: both doravirine and efavirenz were administered with tenofovir disoproxil fumarate (TDF)/emtricitabine (FTC).
Determined by investigator to be related to blinded therapy or to open label TDF/FTC.
Reported by ≥10% participants in any of these groups. AE, adverse event.
Discussion
Doravirine 100 mg in combination with TDF/FTC demonstrated similar antiretroviral efficacy to efavirenz and was associated with a significantly lower incidence of CNS AEs in ART-naive adults with HIV-1 infection. In Part I of the trial, all studied doses of doravirine were generally well tolerated and demonstrated antiretroviral activity and immunological effect similar to efavirenz through 24 weeks of treatment. Therefore, the selection of the dose for further study included additional considerations, such as maintaining therapeutic concentrations of doravirine in the presence of CYP3A inducers or following a missed dose, and providing coverage of a variety of NNRTI mutations, including K103N, Y181C and G190A mutations and the dual K103N/Y181C mutations [10,26]. Based on all available data, doravirine 100 mg once daily was selected for Part II of the trial and the Phase III clinical programme.
In all participants who received the selected dose, doravirine 100 mg once daily demonstrated antiretroviral activity similar to that of efavirenz. In both treatment groups, the proportion of participants with HIV-1 RNA <40 copies/ml increased through week 24 and was maintained up to week 96. The efficacy observed in participants with baseline HIV-1 RNA >100,000 copies/ml was numerically lower for doravirine 100 mg than for efavirenz at week 96; however, this difference should be interpreted in the context of the relatively small subgroup size (32 receiving doravirine 100 mg and 29 receiving efavirenz). In ongoing Phase III trials, doravirine 100 mg has demonstrated similar efficacy to the comparator regimens among participants with baseline HIV-1 RNA >100,000 copies/ml [27,28]. In this trial, doravirine 100 mg also demonstrated an immunological effect similar to that of efavirenz, as measured by the change from baseline in CD4+ T-cell counts.
Doravirine 100 mg was generally safe and well tolerated in this trial. In the key safety analyses, the proportion of participants who experienced CNS AEs was significantly lower for doravirine 100 mg compared with efavirenz at weeks 8 and 24. Drug-related AEs were also less common with doravirine compared with efavirenz, driven primarily by lower rates of drug-related CNS AEs, such as dizziness and abnormal dreams, in the doravirine group. The favourable CNS profile of doravirine versus efavirenz has been confirmed by the week 48 results of the Phase III DRIVE-AHEAD trial, in which the fixed-dose combination of doravirine/lamivudine (3TC)/TDF was compared with the fixed-dose combination of efavirenz/FTC/TDF [28]. Treatment-emergent Grade 2 creatinine elevations were more common in the overall doravirine group (all dose groups combined) than in the efavirenz group; however, this finding was not seen in either of the Phase III trials, where the incidence of Grade 2 creatinine elevations was low in the doravirine groups (2.6% and 2.2%, respectively) and similar to the comparator groups (4.2% and 1.4%, respectively) [29].
Across all treatment groups, the rate of participant discontinuation was relatively high. For example, in Part I, 19% of the participants discontinued due to protocol violations, loss to follow-up, non-compliance or withdrawal by subject. The high discontinuation rate partially accounts for the relatively low response rates observed compared with other Phase II trials with efavirenz [30–32], particularly in Part I, in which 64% of participants in the efavirenz group achieved HIV-1 RNA <40 copies/ml at week 24. The relatively complicated dosing strategy and high pill burden may partially explain the high discontinuation rate; the trial required twice-daily dosing, including both active and placebo tablets to maintain blinding (a total of six tablets per day before and three tablets per day after dose selection). A further limitation was the relatively small size of the treatment groups precluding a formal non-inferiority comparison of efficacy. However, the subsequent Phase III DRIVE-AHEAD trial was adequately powered for statistical comparisons, lacked the high pill burden of this trial, and demonstrated the non-inferior efficacy of doravirine/3TC/TDF versus efavirenz/FTC/TDF at week 48, with 84.3% and 80.8%, respectively, achieving HIV-1 RNA <50 copies/ml [28].
The trial population was primarily young male participants with high baseline CD4+ T-cell counts, potentially limiting the applicability of these results to a more diverse patient population. Ongoing Phase III trials of doravirine are composed of larger, more diverse study populations and greater opportunity to evaluate the relationships between demographics or baseline characteristics and the efficacy and safety of doravirine.
Overall, this trial showed promising results for doravirine as initial treatment of adults with HIV-1 infection. The superior neuropsychiatric profile of doravirine compared with efavirenz and the once-daily dosing regimen are likely to improve treatment adherence [33]. An important outcome was the selection of doravirine 100 mg as the dose for further clinical development in the Phase III trials. The 48-week results of the Phase III trials are favourable, with doravirine showing non-inferior efficacy to ritonavir-boosted darunavir in the DRIVE-FORWARD trial and to efavirenz in the DRIVE-AHEAD trial [27,28], supporting the recent approval of doravirine in the US [34,35], Europe [36,37] and elsewhere. Doravirine has the potential to provide another convenient treatment option that offers efficacy, good tolerability and safety for people living with HIV.
Footnotes
Acknowledgements
We thank all the study participants, and the staff of all the centres taking part in the study. Medical writing assistance, under the direction of the authors, was provided by Edward Rochford and Annette Smith of CMC AFFINITY, a division of McCann Health Medical Communications Ltd, Macclesfield, UK, and Kim Strohmaier of Merck & Co., Inc., Kenilworth, NJ, USA, in accordance with Good Publication Practice (GPP3) guidelines. This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
JMG, JOM-R, DPH, MT, KA, C Hoffmann, FR, OO, RD, AP, DES, JP and SR enrolled the study participants and collected the study data. C Harvey, SK, CF, C Hwang and HT contributed to the coordination and oversight of the study. XX, HW and AR performed the statistical analysis. All authors participated in data interpretation, contributed to the drafting and critical review of the article, and approved the final version of the manuscript.
Funding for this research was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. C Harvey, XX, SK, CF, HW, AR, C Hwang and HT are current or former employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and may own stock and/or stock options in Merck & Co., Inc., Kenilworth, NJ, USA.
Up to 30 April 2018, JMG received honoraria for speaking and advisory boards, and research grants to his institution from ViiV Healthcare, Gilead Sciences, MSD and Janssen. From 1 May 2018, he is an employee (Senior Global Medical Director) of ViiV Healthcare. JOM-R has received honoraria for advisory panels and lectures from MSD, Bristol-Myers Squibb, ViiV Healthcare and AbbVie; and research grants from MSD, Bristol-Myers Squibb, GeneOne, Sanofi Pasteur, GlaxoSmithKline, Sangamo Therapeutics and Gilead Sciences. DPH has received honoraria for speaker's bureau and advisory boards for Merck & Co., Inc., Kenilworth, NJ, USA, Gilead Sciences, and Bristol-Myers Squibb. MT has received research support to AIDS Research Consortium of Atlanta from Bristol-Myers Squibb, CytoDyn Inc., GlaxoSmithKline, Gilead Sciences, MSD, Roche Laboratories, TaiMed Biologics Inc. and ViiV Healthcare. KA has received honoraria from lectures and advisory boards for MSD, Bristol-Myers Squibb, GSK/ViiV Healthcare and Boehringer Ingelheim. C Hoffmann has received honoraria for lecturing, advisory boards, and/or travelling grants from AbbVie, Bristol-Myers Squibb, Boehringer Ingelheim, Gilead Sciences, Hexal, Janssen, MSD and ViiV Healthcare; and research grants from AbbVie, Janssen and Gilead Sciences. FR has received research funding or honoraria from, or consulted for, AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, MSD and ViiV Healthcare. OO has received research grants from Gilead Sciences and Forest Laboratories, and honoraria for advisory boards from Gilead Sciences, Bristol-Myers Squibb, ViiV Healthcare and Janssen. RD owns stock in Merck & Co., Inc., Kenilworth, NJ, USA. AP has received research and/or congress sponsoring, and/or travelling grants from AbbVie, Bristol-Myers Squibb, Gilead Sciences, Hexal, Janssen, MSD and ViiV Healthcare. DES has received travel sponsorship and research grant support from MSD, ViiV Healthcare and Gilead Sciences. JP has received research and/or congress sponsoring, and/or travelling grants from AbbVie, Gilead Sciences, Janssen, MSD and ViiV Healthcare. SR has no conflicts of interest to declare.
Additional file 3: A figure showing participant flow through the study at week 96: Part I and Part II can be found at ![]()
Additional file 4: A table showing proportion of participants with HIV-1 RNA <40 copies/ml at week 24 (Part I) can be found at ![]()
Additional file 5: A table showing most common laboratory changes, occurring in ≥4 participants in any treatment group with indicated grade (based on DAIDS toxicity criteria
a
) that was also an increase from baseline, through week 24 (Part I) can be found at ![]()
Additional file 6: A table showing proportion of participants with HIV-1 RNA <40 copies/ml at weeks 24 and 96 (Parts I+II) can be found at ![]()