
Abstract
Background
Protease inhibitors form the main component of second-line antiretroviral treatment in South Africa. Despite their efficacy, mutations arising within the HIV-1 gag and protease coding regions contribute to the development of resistance against this class of drug. In this paper we investigate a South African HIV-1 subtype C Gag-protease that contains a hinge region mutation and insertion (N37T↑V).
Methods
In vitro single-cycle drug susceptibility and viral replication capacity assays were performed on W1201i, a wild-type reference isolate (MJ4) and a chimeric construct (MJ4GagN37T↑VPR). Additionally, enzyme assays were performed on the N37T↑V protease and a wild-type reference protease.
Results
W1201i showed a small (threefold), but significant (P<0.0001) reduction in drug susceptibility to darunavir compared with MJ4. Substitution of W1201i-Gag with MJ4-Gag resulted in an additional small (twofold), but significant (P<0.01) reduction in susceptibility to lopinavir and atazanavir. The W1201i pseudovirus had a significantly (P<0.01) reduced replication capacity (16.4%) compared with the MJ4. However, this was dramatically increased to 164% (P<0.05) when W1201i-Gag was substituted with MJ4-Gag. Furthermore, the N37T↑V protease displayed reduced catalytic processing compared with the SK154 protease.
Conclusions
Collectively, these data suggest that the N37T↑V mutation and insertion increases viral infectivity and decreases drug susceptibility. These variations are classified as secondary mutations, and indirectly impact inhibitor binding, enzyme fitness and enzyme stability. Additionally, polymorphisms arising in Gag can modify the impact of protease with regards to viral replication and susceptibility to protease inhibitors.
Introduction
HIV-1 subtype C is responsible for the majority of HIV infections in southern Africa, the epicentre of the global HIV pandemic [1]. The HIV-1 protease remains an attractive drug target because it is among the three viral enzymes involved in viral replication and is responsible for the maturation of an infective virion [2,3]. There are currently nine Food and Drug Administration (FDA)-approved protease inhibitors (PIs) available to those infected with HIV [4,5].
PIs are substrate transition state analogues that bind competitively to the protease active site [3]. PIs are prescribed as part of the second-line regimen in South Africa, after failure on a non-nucleoside reverse transcriptase inhibitor (NNRTI)-based regimen [6]. Of the nine approved PIs, lopinavir (LPV) or atazanavir (ATV) are preferred. Darunavir (DRV) is exclusively prescribed for salvage therapy after failure on a PI-based second-line regimen [7]. ATV and LPV are coadministered with ritonavir (RTV) as it functions as a potent inhibitor of the xenobiotic detoxification enzyme cytochrome P450 isoform 3A4, thereby increasing PI bioavailability [8]. However, RTV is avoided as a standalone drug due to the severity of its side effects at higher concentrations, and due to the selection of drug resistance mutations [8–10].
The development of antiretroviral (ARV) drug resistance remains one of the most significant hurdles in the fight for sustained viral suppression within HIV-1-infected patients on ARV therapy (ART) [11]. PI-resistance is mediated by primary mutations located within the active site of the enzyme. These mutations ordinarily give rise to low levels of resistance when not enhanced by secondary mutations distal to the active site. Furthermore, studies indicate that the drug resistance profile of HIV-1 protease is enhanced when compensatory changes arise within the Gag polyprotein, which can modulate the replicative capacity of the virus to further decrease the likelihood of successful treatment [8,12–16].
A recent study has reported on an HIV-1 isolate (W1201i) from an HIV-1-infected South African drug-naive infant with an N37T↑V hinge region mutation and insertion in the protease enzyme [17]. Insertion mutations in protease are rarely reported, and their impact on drug susceptibility has not been well characterized [18]. The hinge (residues 35–42 and 57–61) of protease is highly variable and controls the molecular dynamics (MD) of the flap region (residues 46–54) [19]. The flap region, in turn, controls substrate entry into the active site and influences drug binding [8]. MD simulations suggest that the N37T↑V hinge region mutation increases the dynamics of the flap region which alters its kinetic profile [20]. The altered kinetics of the N37T↑V protease may result in reduced PI drug susceptibility [8,20,21]. Here, we report on the in vitro drug susceptibility, viral replication capacity (RC) and enzymatic catalytic efficiency of the W1201i isolate.
Methods
Cohort study
Plasma samples from newly diagnosed PI-naive children younger than 2 years of age were investigated in a previous study that examined drug resistance following short-course treatment to prevent mother-to-child HIV transmission [17]. A sample from an infant (W1201i, GenBank: KX902482.1) was selected for further study because the isolate was shown to contain a protease hinge region mutation and insertion (N37T↑V). We chose to name the protease hinge region mutation and insertion as N37T↑V. The N37T↑V protease also contains other notable polymorphisms in addition to the hinge region substitution and insertion (Figure 1).

Homology model of the N37T↑V protease (derived from W1201i)
Prenatally, the mother of W1201i was PI-naive but had been exposed to nevirapine (NVP) for 42 days before labour to prevent mother-to-child transmission. The infant had been treated with azidothymidine (AZT) as prophylaxis after birth and was PI-naive at the time blood samples were taken [17].
Vector construction
Total viral RNA was extracted from patient plasma using the QIAmp Viral RNA Mini kit (QIAGEN, Antwerp, Belgium) and reverse transcribed using a Thermoscript RT-PCR kit (Invitrogen, CA, USA). The 1.8 kb gag-protease amplicon was amplified by nested polymerase chain reaction (PCR) using an Expand High Fidelity PCR kit (Roche Applied Science, Basel, Switzerland). Population-based Sanger sequencing of gag-protease and the construction of the patient-derived HIV-1 expression vector was performed as previously described by Giandhari et al. [22]. Briefly, the patient-derived HIV-1 expression vector (W1201i) was constructed by cloning the ±1.8 kb gag-protease amplicon into the p8.9NSX+ HIV-1 expression vector, which contains a NotI site at the beginning of gag (HXB2 position 772) and an XhoI site at the end of the protease coding region (HXB2 position 2563) [22]. Furthermore, an artificial chimeric construct was created by exchanging the patient-derived gag from W1201i with the gag from an HIV-1 subtype C wild-type reference isolate (MJ4, GenBank: AF321523.1) to generate MJ4GagN37T↑V. This was achieved through restriction with NotI and a unique ApaI site in the p7 nucleocapsid coding region of both the p8.9MJ4GFP wild-type reference vector and the W1201i vector. A multi-drug resistant reference control was included that contained six PI resistance mutations (that is, L10I, K20R, M46I, I54V, I62V and V82A) and five background mutations (that is, E35D, S39P, Q61H, T74S and I77V), compared with the MJ4 wild-type protease sequence [22,23].
Therefore, four p8.9NSX+-based constructs were ultimately used for the drug susceptibility and RC experiments; namely, MJ4, resistance control, W1201i (full Gag-protease from infant) and MJ4GagN37T↑VPR (consisting of Gag derived from MJ4 and protease derived from W1201i). The W1201i gag-protease, chimeric construct and the resistance control were each cloned into a p8.9NSX+ expression vector to use in the single-cycle phenotypic assays.
The viral vectors contain only the core HIV proteins (Gag, protease, reverse transcriptase and integrase) and are HIV envelope protein-deficient (Additional file 1). Two additional vectors were used in this study; namely, pMDG and pCSFLW. The pMDG vector encodes the vesicular stomatitis virus G protein which associates with envelope-deficient HIV-1 core proteins to form an infective pseudotype virus. The pCSFLW vector encodes the firefly luciferase reporter gene inserted between HIV-1 3’ and 5’ long terminal repeats. The expression of the integrated luciferase gene allows the direct measurement of infectivity via bioluminescence [24].
Phenotypic drug susceptibility
A single-cycle non-replication assay was used for phenotypic drug susceptibility testing [24]. Briefly, HEK293T cells were transfected with the HIV-1 expression vector, the pMDG vector that expresses vesicular stomatitis virus protein G for entry, and the pCSFLW vector that encodes the firefly luciferase reporter gene. The transfected cells were harvested after incubation at 37°C under 5% CO2 for 36 h. The harvested cells were seeded in 96-well culture plates with threefold serially diluted PIs. After 24 h, the supernatants were transferred to the corresponding wells of an indicator plate that contained uninfected HEK293T cells. The degree of pseudoviral infection was measured 48 h post-transfer, as determined by the expression of firefly luciferase in infected target cells. Luciferin-containing Bright-Glo (Promega, CA, USA), was added to each well and incubated for 2 min before reading the luminescence on a Victor 3 Luminometer (Perkin Elmer, MA, USA).
The half maximal inhibitory concentration (IC50) values were calculated for each sample and drug (DRV, ATV and LPV). The drug susceptibility (expressed as fold change [FC]) of W1201i, the resistance control and the MJ4GagN37T↑VPR construct was expressed relative to MJ4GP. The lower biological cutoff value for each drug was set at the 99th percentile of IC50 replicates for MJ4: LPV (1.2 FC), ATV (1.4 FC) and DRV (1.7 FC). Values above these levels indicate a decrease in drug susceptibility. Figures and statistics were compiled in GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA).
Viral RC
RC was determined by harvesting pseudovirions from transfected drug-free HEK293T cells and infecting fresh HEK293T cells with the neat viral stocks. The subsequent expression of firefly luciferase was quantified 48 h later, as described in Phenotypic drug susceptibility in Methods. Input virus was quantified using a chemiluminescent p24-antigen ELISA assay (Protocol 2; Aalto Bio Reagents Ltd., Dublin, Ireland). The RC was calculated by referring to the ratio of input virus (nanogram p24) against the level of luciferase expression.
Protease characterization
The N37T↑V protease derived from the W1201i isolate was purified as a recombinant protein through immobilized metal ion affinity chromatography [25]. A protease derived from a different wild-type reference isolate (SK154, GenBank: HM593163.1) was used as a control for the enzymatic characterization of N37T↑V. It should be noted that the SK154 protease was used only as a wild-type control for the enzymatic characterization of N37T↑V. MJ4 was utilized as a control for drug susceptibility and RC experiments. The SK154 protease was purified as described by Naicker et al. [19].
A Q7K mutation, known to decrease autolysis without affecting catalysis, was incorporated into the protease coding region of both SK154 and N37T↑V. Although the Q7K mutation is in close proximity to the active site, there is no evidence that it affects the activity of the protease [26]. This mutation was, therefore, not incorporated into the protease (N37T↑V) coding region of W1201i for phenotypic drug susceptibility assessment.
Enzymatic parameters were determined following the hydrolysis of the HIV-1 protease fluorogenic substrate (Abz-Arg-Val-Nle-Phe(NO2)-Glu-Ala-Nle-NH2), which mimics the CA/p2 cleavage site in the HIV-1 Gag poly-protein. The Nle-Phe(NO2) residue efficiently quenches the substrate aminobenzoyl (Abz) group. Quenching is abolished when the substrate is cleaved, which allows Abz to fluoresce at 425 nm when excited at 337 nm [27,28]. The fluorescence emission measurements of all samples were recorded for 1 min using an excitation bandwidth of 2.5 nm and an emission bandwidth of 5 nm. The complete cleavage of 1 nmol substrate was measured and used to convert the emission intensity of the subsequent experiments to activity.
To measure specific activity, the fluorogenic substrate concentration was kept constant at 50 μM. HIV protease concentration was varied for each assay (10 to 50 nM). The reaction velocity (μmol•min−1) was plotted against the amount of protease in milligrams. The specific activity (μmol•min−1•mg−1) was calculated from the gradient of the resultant curve. The catalytic efficiency (kcat/KM) was determined with a variable substrate concentration (1 to 10 μM) and a constant protease concentration (50 nM). The catalytic constant (kcat) for each protease was calculated from their respective specific activity values.
Results
Genotypic analysis of gag-protease from W1201i The protease encoding region of the W1201i amplicon was previously shown to contain a mutation and a rare insertion at position 37 in the hinge region [10]. A homology model of the W1201i protease containing the N37T↑V as well as a sequence alignment with the wild-type consensus (SK154), MJ4 and resistance control proteases is shown in Figure 1. In this study, SK154 was used as a control for the enzymatic characterization of N37T↑V. Similarly, MJ4 was used as a control when assaying W1201i for drug susceptibility and RC. Compared with the wild-type (SK154), the variant protease displayed the following mutations: Q7K, I13V, G16E, I36T, N37T↑V, P39S, D60E, Q61E, I62V, L63P, V77I and M89L. Compared to the MJ4, the variant protease displayed the following background mutations: Q7K, I13V, M36T, N37T↑V, Q61E and M89L. Furthermore, three minor ATV resistance mutations were also found (MJ4 versus N37T↑V): G16E, D60E and I62V [29]. Both MJ4-protease and N37T↑V exhibited L63P, a minor LPV resistance mutation. Suñé et al. [30] reported that L63P can have significant effects on viral fitness; however, since MJ4 contains L63P we cannot account for its effects in this study. Mutations were designated as drug resistance mutations if they were cited as such on either the Stanford HIV Drug Resistance Database or the protease drug resistance mutations list curated by Wensing et al. [29].
Given the close link between Gag and protease, we also sequenced the Gag region of W1201i. All polymorphisms and their locations in Gag are indicated in Figure 2 which shows a sequence alignment between W1201i and MJ4. Genotypic analysis of W1201i gag showed several insertions in, or near, cleavage sites, which could potentially influence drug susceptibility and RC [31,32]. Mutations include a single PTAPP duplication and LE insertion in p6Gag, as well as an I372L↑M mutation and insertion in the p2/NC cleavage site. In addition to I372L↑M, the p2/NC cleavage site showed the following polymorphisms: S369N, S371N, I373M and G377S. Moreover, a previously unreported MSQAG duplication was found between the CA/p2 and p2/NC cleavage sites.

Alignment of the MJ4-Gag sequence and W1201i-Gag
Phenotypic drug susceptibility
The W1201i sample-derived pseudovirus was susceptible to both LPV (FC 2.0) and ATV (FC 1.6), and no significant difference (P>0.05) was observed compared with the MJ4 control (Figure 3). However, a reduced susceptibility was observed to DRV (FC 4.6), which was significantly higher (P<0.0001) than MJ4. With the substitution of W1201i-Gag, MJ4GagN37T↑VPR showed a small, but significant (P<0.001), reduction in susceptibility for both LPV (FC 3.4) and ATV (FC 3.7). No further significant reduction (P>0.05) was observed for DRV (FC 4.7). The resistance control exhibited a significant (P<0.0001) reduction in susceptibility (FC>10) towards all three PIs.

The phenotypic drug susceptibility of four pseudoviruses
Viral RC
When assessed for RC, the W1201i pseudovirus showed a significant (P<0.05) reduction in RC (16.4%), compared with MJ4 (100%; Figure 4). However, with the substitution of the Gag, the resulting MJ4GagN37T↑VPR pseudovirus showed a drastic increase in RC (164%; P<0.01) compared with MJ4, suggesting that the polymorphisms in the W1201i Gag caused the observed reduction in RC.
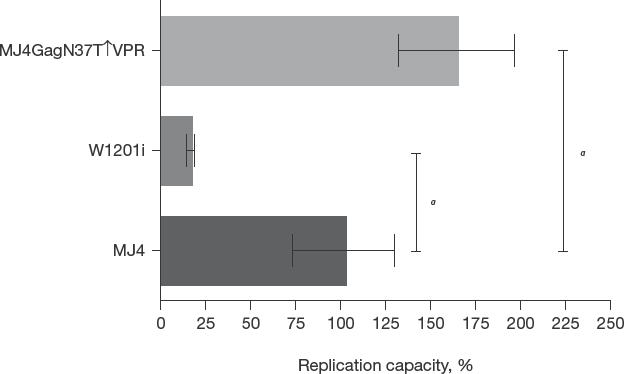
Replication capacity of the isolates/constructs
Protease characterization
The enzyme kinetic parameters of the wild-type (SK154) and N37T↑V proteases were determined via fluorogenic substrate cleavage (Table 1). The specific activity was found to be 12 ±1 μmol•min−1•mg−1 for wild-type and 1.8 ±0.2 μmol•min−1•mg−1 for N37T↑V, indicating that the variant shows reduced specificity toward the fluorogenic substrate. The catalytic constant (substrate turnover per second) was determined from the specific activity data and was found to be 6.4 ±0.2 and 1.5 ±0.2 s−1 for wild-type and N37T↑V, respectively. The wild-type protease was five times more catalytically efficient than the N37T↑V variant with a kcat/KM value of 2.4 ±0.1 s−1•μM−1 compared with 0.5 ± 0.1 s−1•μM−1 for N37T↑V. Cumulatively, the data indicate that the N37TV variant processes the fluorogenic substrate less efficiently.
Enzymatic parameters of the wild-type subtype C and N37T↑V proteases (mean ±sd)

SK154, GenBank: HM593163.1. See Genotypic analysis of gag-protease from W1201i in Results.
Discussion
It has been reported that insertion mutations can modulate the activity of the protease enzyme and impact on viral RC [15,18,33,34]. Nucleotide insertions occur most commonly between codon 32 and 42 in the protease coding region [18]. Insertion mutations are rare and their prevalence ranges from 0.1 to 4.55% depending on the region, subtype and codon type [15,35,36]. Unfortunately, insertion mutations are neither curated in the Stanford or Los Alamos databases, and information on their prevalence per subtype is not currently available. The highest prevalence of insertion mutations (4.55%) was reported in Hong Kong. However, these were subtype B isolates, and only codon 35 was analysed. Pereira-Vaz et al. [35] found that the prevalence of codon 35 insertions in subtype C isolates from Portugal were 2.69%. In a previous study we have shown that the N37T↑V mutation and insertion increases the molecular dynamics of both the flap and the hinge regions in HIV-1 protease [20]. Here, we investigated the in vitro phenotypic drug susceptibility, RC and enzymatic properties of this insertion variant to understand its impact on HIV-1 protease.
As anticipated, W1201i displayed a significant reduction in RC compared with the MJ4 control pseudovirus. However, when the W1201i Gag was substituted with MJ4 Gag, a significant increase in RC, relative to the MJ4 control, was observed. Thus, N37T↑V appears to catalyse the cleavage of MJ4-Gag more effectively. We postulate that the selection of N37T↑V is dependent on the variations within Gag, and not vice versa since natural selection would favour a virus with an increased RC. However, multiple clinical trials have shown that decreased RCs could be advantageous, as such viruses experience longer survival times and increased population prevalence compared with viruses with increased fitness and disease progression [37]. However, a limitation of this study is the use of RC as a measure for replication fitness. Although RC reflects on the potential replication fitness of a virus and is typically reported by commercial phenotypic drug susceptibility assays (that is, Monogram Biosciences) it is not a definitive measure of fitness.
In the context of its native Gag, a small, but statistically significant decrease in susceptibility to DRV was observed, while RC was significantly impaired. However, in the presence of the unrelated MJ4-Gag, a small but significant decrease in susceptibility was observed to all three PIs while RC was significantly improved. Cumulatively, the data show that the W1201i Gag confers no advantage to RC and PI susceptibility. However, in the absence of a wild-type baseline sample for this particular patient, a comparison was made to an unrelated wild-type reference gag-protease (that is, MJ4) as an alternative. We have identified several differences (that is, background mutations) in the amino acid sequences between MJ4 and W1201i, and it is possible that these could have influenced the phenotypic results observed for W1201i.
Gag genotyping revealed notable polymorphisms and insertions. The W1201i Gag displayed a PTAPP duplication and variations in the p2/NC cleavage site (S369N, S371N, I372L↑M, I373M and G377S). Gag insertion variants are usually co-selected with other polymorphisms to help maintain viral fitness [38]. However, these polymorphisms can accumulate in the absence of drug pressures [39]. It has been shown that nucleoside-based ART positively correlates with PTAP duplications in subtype B strains [39]. However, the PTAP duplication occurs at a higher frequency in subtype C variants [39,40]. A study performed by Martins et al. [32] concluded that PTAP duplications had little effect on the viral infectivity in wild-type strains, but only led to increased viral infectivity, and decreased drug susceptibility, when modified by specific drug resistance mutations. Our results are in agreement with the findings of Martins and colleagues. The W1201i Gag did not increase RC; rather, W1201i Gag appears to decrease the viral RC. Similarly, it has been reported that p2/NC cleavage site mutations will accumulate under PI pressure [41]. It is currently believed that cleavage site mutations accumulate to restore lost viral fitness due to the development of mutations within protease [42]. Our results do not corroborate these findings because neither the PTAP duplication nor the cleavage site mutations were able to restore the RC of W1201i. However, we did not investigate the RC of W1201i in the absence of these polymorphisms.
The W1201i isolate presented a previously unreported MSQAG insertion in proximity to both the CA/p2 and p2/NC cleavage sites (Figure 2). Tamiya et al. [14] suggested that insertions near the CA/p2 cleavage may compromise RC; however, the mechanism has not been established. It is believed that insertions alter the conformation of the cleavage site and limit access to the protease enzyme. It is reasonable to expect that insertions could impact RC, as the cleavage of Gag is thought to be controlled by the shape of the cleavage site and not a particular amino acid sequence [43]. Therefore, it is possible that the observed reduction in the RC of W1201i is due to a deleterious effect conferred by the MSQAG insertion. Moreover, these data are in agreement with the low specific activity, catalytic turnover and catalytic efficiency displayed by the N37T↑V protease towards a substrate mimicking the CA/p2 cleavage site. If the N37T↑V protease displays lowered catalytic efficiency toward the substrate, then further structural changes such as an MSQAG insertion could possibly enhance the detrimental effect on specificity.
The fold change of susceptibility to LPV, ATV and DRV was analysed (Figure 3A–3C). W1201i displayed a minimal, but statistically significant decrease in drug susceptibility to ATV, whereas the chimeric construct displayed a minimal but statistically significant decrease in drug susceptibility to LPV, ATV and DRV. Under normal conditions, protease insertion variants are usually fully susceptible to all PIs [34]. However, here the variant is modified by only three minor ATV resistance mutations, compared with MJ4. Despite this, the N37T↑V protease displayed decreased susceptibility to both LPV and DRV. Considering the drug resistance profile of N37T↑V, it is possible that the observed reduction in susceptibility to LPV and DRV is due to the presence of the hinge region insertion and mutation. It is unclear to what extent background mutations in PR, or polymorphisms and insertions in Gag, could have contributed to the observed phenotype and RC of W1201i. Future studies will unravel the role of these potentially confounding factors by screening additional chimeric constructs and assessing the impact of the observed changes in PR on viral fitness in competitive viral replication assays.
Exposure to ARVs can alter the genetic profile of HIV-1. For this reason, it is advantageous to know the ART history of the cohort participant from whom samples were taken. To our knowledge, neither the mother nor the infant have received PI-based treatment prior to the sampling point, and although PI-exposure would have been highly unlikely, the possibility cannot be excluded. The sharing of ARV drugs is not uncommon in the setting of a low-to-middle income country [44,45], and it is possible that the patient could have obtained PIs from a family member or friend without reporting this to her medical service provider. However, taking into consideration that the patient ‘developed’ this peculiar and complex insertion mutation in the absence of any other PI mutations may suggest that an alternative mechanism, apart from ART-induced resistance selection, may have been at play. It is well-known that the immune system imposes immense pressure on the virus to escape, and as an evolutionary consequence of this, the virus introduces various modifications to its proteins, including Gag and protease to escape the immune response [46–48]. Regarding the development of the insertion mutation in the W1201i patient, it is therefore possible that immune pressure could have led directly to the selection of the insertion mutations in the protease. Alternatively, immune pressure could have selected for the various polymorphisms and insertions observed in the patient's gag gene, which subsequently could have instigated the change in protease. The exact cause for the selection of the insertion mutation in the protease, however, remains speculative.
In this study, we have once again shown a clear link between protease and Gag based on phenotypic testing. A body of evidence is accumulating in support of the fact that Gag polymorphisms can modulate PI susceptibility, even in the absence of protease drug resistance mutations [22,24,48,49]. Despite the evidence, genotypic testing is still focused predominantly on the protease sequence [49].
Footnotes
Acknowledgements
The research reported in this publication was supported by the South African Medical Research Council under a Self-Initiated Research Grant to YS. The views and opinions expressed are those of the authors and do not necessarily represent the official views of the SAMRC. This work was supported by the University of the Witwatersrand, the South African National Research Foundation Grant 68898 (HWD), and the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation Grant 64788 (HWD). ATV, DRV and LPV were supplied by the National Institutes of Health (NIH) AIDS Reagent and Reference Program. Plasmids p8.9 and pMDG were supplied by Didier Trono (École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland), and plasmid pCSFLW was supplied by Nigel Temper-ton (University College London, London, United Kingdom). We thank Johanna Ledwaba and Gillian Hunt for identifying this rare protease sequence and Jennifer Giandhari (all affiliated with Centre for HIV and STIs, National Institute for Communicable Diseases [NICD] of the National Health Laboratory Service [NHLS], Johannesburg, South Africa) for help with gag-protease sequencing and providing the protocols used to test drug susceptibility and viral infectivity.
The authors declare no competing interests.