
Abstract
Background
Cabotegravir (CAB) is an integrase strand transfer inhibitor in development as a long-acting injectable formulation, with an oral formulation used during a safety lead-in period. Tuberculosis (TB)–HIV coinfection is common, often requiring simultaneous treatment. Rifabutin (RBT) is an alternative antimycobacterial agent for TB and a moderate inducer of cytochrome P450 and UDP-glucuronosyltransferase isoenzymes. This study evaluated the impact of RBT on the pharmacokinetics (PK) of oral CAB.
Methods
In this Phase I, single-centre, open-label, two-period, fixed-sequence, drug interaction study, subjects received oral CAB 30 mg once daily for 14 days in period 1, and oral CAB plus RBT 300 mg once daily for 14 days in period 2. Serial PK sampling was performed on days 14 and 28. Geometric least squares (GLS) mean ratios with associated 90% CIs were calculated to compare CAB non-compartmental PK parameters following CAB+RBT versus CAB alone. Safety was also assessed.
Results
A total of 15 male subjects were enrolled and 12 completed all treatments. Comparing CAB+RBT with CAB alone, the GLS mean ratios (90% CIs) for CAB area under the concentration–time curve from time zero to the end of the dosing interval (AUC0–τ), maximum observed plasma concentration (Cmax) and concentration at the end of the dosing interval (Cτ) were 0.79 (0.74, 0.83), 0.83 (0.76, 0.90) and 0.74 (0.70, 0.78), respectively. 11 subjects reported 24 adverse events (AEs); 22 were reported with CAB+RBT (3 drug-related) and 2 with CAB alone (not drug-related). All AEs resolved by study end.
Conclusions
RBT had a modest impact on plasma CAB exposure following oral coadministration, resulting in overall plasma CAB trough exposures above the 10 mg oral dose shown to maintain viral suppression in HIV-1-infected subjects. Oral CAB can be coadministered with RBT without dosage adjustment.
Introduction
Tuberculosis (TB)–HIV coinfection is common and both conditions often need to be treated simultaneously. TB–HIV coinfection guidelines provide recommendations for safe and effective anti-TB treatments and for their concurrent use with antiretroviral therapy (ART) [1]. However, treatment for HIV and TB is often complicated by potential drug–drug interactions (DDIs). Compounds like rifampin (RIF), the most widely-used rifamycin for TB treatment, can significantly alter the pharmacokinetics (PK) of coadministered antiretroviral (ARV) agents owing to potent induction of major cytochrome P450 (CYP) 3A and UDP-glucuronosyltransferase (UGT) enzymes [2–5]. Consequently, several classes of ARV agents, including protease inhibitors, non-nucleoside reverse transcriptase inhibitors and some integrase inhibitors are contraindicated for use with RIF. Rifabutin (RBT) is chemically related to RIF and is considered a weaker enzyme inducer of UGTs and CYP3A compared with RIF [6,7]. Consequently, RBT is often used as an alternative antimycobacterial agent in the treatment of Mycobacterium tuberculosis (TB) and, in some cases, for latent TB infection as well as for both treatment and prevention of Mycobacterium avium complex (MAC) in advanced HIV infection [8–10].
Cabotegravir (CAB) is an integrase strand transfer inhibitor (INSTI) in development for the treatment and prevention of HIV-1 infection [11]. CAB has been formulated as a long-acting injectable suspension (CAB LA) for intramuscular administration and has a favourable PK profile supporting monthly or every 2-month administration [12], providing a viable alternative in scenarios where adherence to daily oral ART might be challenging. An immediate-release oral formulation of CAB 30 mg has been selected as the oral lead-in dose as a safety check prior to initiating CAB LA injections. Maintenance of HIV suppression has been demonstrated with a 2-drug regimen of CAB LA + rilpivirine (RPV) LA, administered as separate intramuscular (IM) injections every 4 or 8 weeks [13]. CAB LA is also being developed as a single agent to be used for pre-exposure prophylaxis (PrEP) [11].
CAB is metabolized primarily through UGT1A1 with a minor contribution of UGT1A9 [14] and has a terminal elimination phase half-life (t1/2) of 40 h [15]. CAB does not induce or inhibit metabolic enzymes and many transporters, and thus has a low propensity to be a victim or perpetrator of drug interactions. The pathways of CAB metabolism and elimination are similar despite different routes of administration. This may provide a useful model for imputation of predicted and observed DDIs with the oral tablet to the long-acting formulation [16,17]. A previous clinical study demonstrated a significant decrease in the concentration of CAB when co-administered with RIF, consistent with predictions from a physiological-based PK analysis of CAB and RIF. Therefore, coadministration of RIF with either the oral or LA formulations of CAB is not recommended without further study [18,19]. This one-way drug interaction study was conducted to determine the effects of RBT administration on the PK of oral CAB in healthy adults.
Methods
Study design
This was a Phase I, single-centre, open-label, fixed-sequence, two-period study conducted in healthy adults to evaluate the effect of RBT on the steady-state PK of oral CAB (NCT03149848; Figure 1). All subjects underwent a screening visit within 30 days from the first dose of study drug. In period 1, subjects received oral CAB 30 mg once daily for 14 days. In period 2, CAB dosing was continued and RBT 300 mg (2x 150 mg capsules) once daily was added for 14 days. There was no washout between period 1 and period 2. Subjects completed a follow-up visit approximately 10–14 days after the last dose of study treatment.

Study design
CAB 30 mg was administered on days 1, 13 and 14, and the combination of CAB 30 mg and RBT 300 mg was administered on days 15, 21, 26, 27 and 28, by authorized study personnel in the unit to each subject in the clinic under fasting conditions. On days 2–12, CAB 30 mg was taken at home, and on days 16–20 and 22–25, combination doses were taken at home. During period 1, blood samples to determine CAB PK concentrations were obtained pre-dose on day 13, pre-dose on day 14, and then at 1, 2, 3, 4, 8, 12 and 24 h post-CAB dosing on day 14. During period 2, blood samples to determine CAB PK concentrations were obtained pre-dose on days 26 and 27, pre-dose on day 28, and then at 1, 2, 3, 4, 8, 12 and 24 h post-CAB + RBT dosing on day 28. Dosing on serial PK days (day 14 in period 1 and day 28 in period 2) was after an overnight fast of at least 6 h, followed by dose administration with 240 ml of water, with no food allowed for 4 h after dosing.
Safety assessments included monitoring of adverse events (AEs), clinical laboratory tests, vital signs and electrocardiograms (ECGs). Physical examinations and pregnancy tests were also conducted. An ocular examination assessing signs or symptoms of uveitis was performed only for those subjects who developed eye symptoms. Study staff were responsible for detecting, documenting and reporting events that met the definition of an AE; AE information volunteered by the subject or detected by other means was collected from the start of study treatment until the follow-up visit. All SAEs were recorded and reported to the study sponsor within 24 h.
Study population
Eligible subjects were healthy men and women between 18 and 65 years of age with body weight of ≥50 kg and body mass index of 18.5 to 31.0 kg/m2. Women of childbearing potential were required to be sexually inactive by abstinence or to use highly effective contraceptive methods. Subjects were required to have normal haematology and chemistry results, including alanine aminotransferase (ALT), alkaline phosphatase and bilirubin ≤1.5x upper limit of normal (ULN), and negative screening results for HIV-1, TB, hepatitis B and hepatitis C. Subjects were excluded if they had a current or chronic history of liver disease, known hepatic or biliary abnormalities, a history of clinically significant cardiovascular disease or a history of uveitis. Subjects with a history of regular alcohol consumption in the 6 months prior to the study, current or recent use of tobacco-containing products within 30 days prior to the screening visit, or subjects who required use of prescription or non-prescription drugs, were excluded. Other exclusion criteria included a history of cholecystectomy or other gastrointestinal surgery, peptic ulceration or pancreatitis (within the 6 months preceding screening) or being determined by the investigator to have a high risk of seizures.
Written informed consent was obtained from each subject prior to the performance of any study-specific procedure. This study was conducted in accordance with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Good Clinical Practice, all applicable patient privacy requirements and the ethical principles outlined in the Declaration of Helsinki (2013).
End points and assessments
The primary objective of this study was to compare the steady state PK of CAB 30 mg + RBT 300 mg once daily compared with CAB alone. The primary end points were to determine CAB maximum observed plasma concentration (Cmax) and area under the concentration-time curve from time zero to the end of the dosing interval (AUC0-τ) Secondary end points included plasma CAB concentration at the end of the dosing interval (C7), time to Cmax (tmax), oral clearance (CL/F), AEs, clinical laboratory evaluations, ECGs and vital sign assessments.
Bioanalytical methods
Blood samples were collected via an indwelling cannula (or by direct venipuncture), collected into a 2 ml tripotassium ethylenediaminetetraacetic acid (K3EDTA)-treated tube, and gently inverted 10x before cooling on ice. Within 1 h of collection, the plasma was separated by refrigerated (2-8°C) centrifugation at 1,600 x g for 15 min. Supernatant plasma was transferred into a single 2 ml tube and kept frozen at −20°C until measurement. Following protein precipitation, samples of plasma were extracted by protein precipitation with acetonitrile containing the internal standard, [13C2H15N]-CAB. An aliquot (2-7 μl) was injected onto a Luna C18(2)-HST column (Phenomonex, Torrance, CA, USA) and eluted with a mobile phase (A: 0.4% formic acid in water; B: 25-mM ammonium formate in acetonitrile:water [80:20, v/v]) using the following gradient: 0.0-0.3 min: 40% B; 0.3-2.0 min: 40-90% B; and 2.0-2.5 min: 90% B. A Sciex API-4000 mass spectrometer (AB Sciex, Framingham, MA, USA) with an electrospray source that employed positive-ion mode and multiple-reaction monitoring detected the analytes (CAB transition mass-to-charge ratio, 406.0-263.0; internal standard transition mass-to-charge ratio, 410.1-263.0). CAB plasma samples were processed by Covance Laboratories (Madison, WI, USA). The range of quantification for CAB from a 25-μl plasma sample was 25-25,000 ng/ml. Quality control samples containing CAB at three known concentrations were stored and analysed with each batch of samples against separately prepared calibration standards. For an analysis to be acceptable, no more than one-third of the quality control results could deviate from the nominal concentration by more than 15%, with ≥50% of the results from each sample within 15% of the nominal value.
Statistical methods
The sample size of 15 was chosen to obtain 12 evaluable subjects, based on an expected withdrawal rate of approximately 20%, and the within-subject variability of CAB. Based on a within-subject coefficient of variation of 11.4% and a sample size of 12 evaluable subjects, the lower and upper bounds of the 90% CI for the treatment difference on a logarithmic scale were estimated to occur within 8.3% of the point estimate for AUC0-τ and Cmax.
CAB plasma concentration-time data were analysed by non-compartmental methods based on the actual sampling times with WinNonlin Phoenix 6.3 (Certara Corporation, Princeton, NJ, USA). Plasma PK parameters, which were calculated from plasma concentration-time data, included the following: Cmax, concentration from time zero to the end of the dosing interval (C0-τ), AUC0-τ, and CL/F. PK parameters were log transformed before analysis and geometric least squares (GLS) mean ratios with associated 90% CIs were calculated to compare CAB non-compartmental PK parameters following CAB + RBT versus CAB alone using SAS (version 9.4) PROC MIXED procedure. Safety data were tabulated and summarized descriptively.
Results
Study population
A total of 15 male subjects were enrolled in the study and 12 completed all treatments. The majority of subjects were White (93%) with a mean (standard deviation [
Subject baseline characteristics

BMI, body mass index; CAB, cabotegravir; RBT, rifabutin.
PK Analyses
Comparisons of plasma CAB PK parameters following coadministration with RBT (RBT 300 mg + CAB 30 mg) to CAB 30 mg alone are shown in Table 2 and Figure 2. Median (range) tmax was 3.0 h (1.0-4.0 h) following CAB alone and 2.5 h (1.0-4.0 h) following CAB with RBT. RBT increased CAB CL/F by 27% and reduced CAB AUC0-τ, Cmax and Cτ by 21%, 17% and 26%, respectively; the corresponding GLS mean ratios (90% CIs) for CAB AUC0-τ, Cmax and Cτ were 0.79 (0.74, 0.83), 0.83 (0.76, 0.90) and 0.74 (0.70, 0.78), respectively.
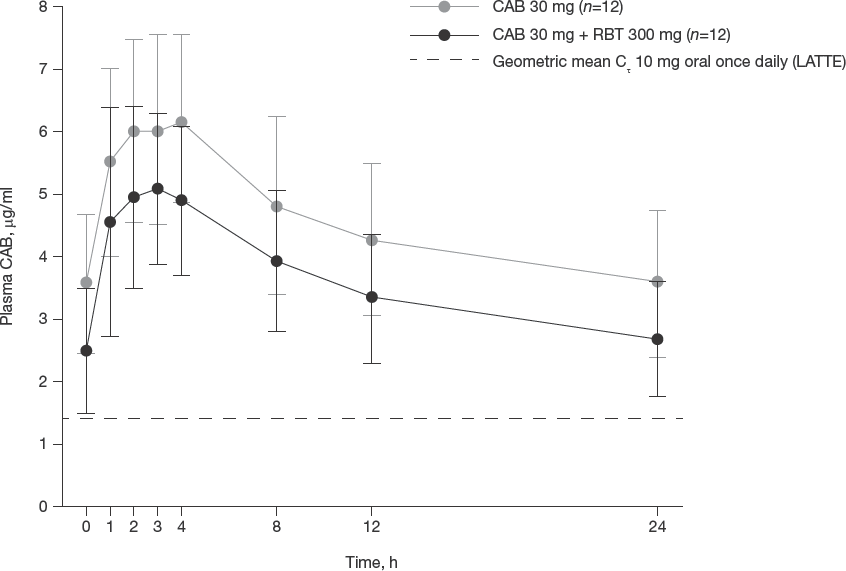
Mean (±
Summary of CAB pharmacokinetic and treatment comparisons
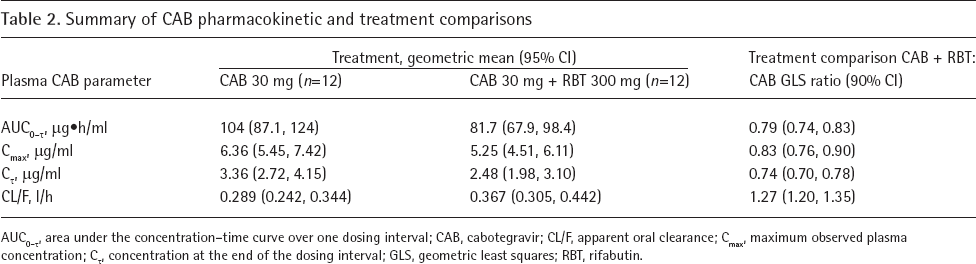
AUC0-τ' area under the concentration-time curve over one dosing interval; CAB, cabotegravir; CL/F, apparent oral clearance; Cmax, maximum observed plasma concentration; Cτ, concentration at the end of the dosing interval; GLS, geometric least squares; RBT, rifabutin.
Safety
A total of 24 AEs were reported by 11 subjects during the on-treatment and follow-up phases of the study (Table 3). The majority of AEs (75% [18/24 events]) were grade 1 and occurred during CAB + RBT coadministration. Three subjects developed AEs that were considered drug-related during the study, one during CAB + RBT coadministration and two in the follow-phase; none were reported during CAB alone administration. One subject developed mild, grade 1 neutropenia and leukopenia and an increase in ALT of grade 3 severity during CAB + RBT coadministration. In the follow-up phase, one subject developed mild neutropenia and another subject developed grade 4 leukopenia. The latter was of grade 1 severity during period 2 (CAB + RBT coadministration) but became of grade 3 severity during the follow-up phase (described in detail below). All events were generally consistent with the known AE profile of RIF and resolved during the study.
Summary of AEs reported during the study

Data are n (%).
In treatment is defined as days 1–28.
Follow-up phase is defined as after day 28, or after treatment stopped, and being followed until discharged from the study. AE, adverse event; ALT, alanine aminotransferase; CAB, cabotegravir; RBT, rifabutin.
One subject developed a serious AE (SAE) during the follow-up period that was considered drug-related by the investigator. During CAB + RBT coadministration, this 55-year-old subject developed a mild, asymptomatic, grade 1 ALT elevation (ALT = 86 IU/1; 1.7x ULN) that fluctuated during period 2 participation. A maximum ALT elevation of a grade 3 intensity (ALT = 417 IU/1; 8.3x ULN) developed during the follow-up period approximately 3 weeks after treatment had ended. An extensive hepatic work-up failed to identify any evidence of a viral aetiology, autoimmune liver disease or Wilson's disease. The subject also had normal immunoglobulins and alpha-1-antitrypsin levels, denied exposure to other drugs or alcohol, and did not report any relevant family history. A liver ultrasound demonstrated hepatic steatosis. The subject remained asymptomatic throughout the entire study and during the follow-up period and the SAE resolved 44 days after date of onset.
Three subjects developed AEs leading to premature study drug withdrawal. In period 1, one subject receiving CAB reached protocol-defined stopping criteria of elevated ALT with values ≥3x ULN and was withdrawn from the study prior to collection of PK samples. The increase in ALT accompanied by a rise in creatine kinase was attributed to strenuous exercise that the subject had undertaken 2 days prior to the day of blood collection, which was prohibited per the study protocol. In period 2, two subjects were withdrawn from the study due to AEs that occurred during CAB + RBT co-administration. One subject developed food poisoning and another participant developed a viral illness. Both subjects had incomplete PK sample collection and were excluded from the PK summary population for analysis due to absence of paired data across both periods. No clinically significant trends were observed in laboratory values, ECGs or vital signs, and no cellular, granular or hyaline casts, or erythrocytes and leukocytes were seen on urinalysis. No ocular-related events occurred.
Discussion
RBT modestly increased CAB CL/F by 27% following repeat dose coadministration, which resulted in a decrease in plasma CAB AUC0–τ, Cmax and Cτ by 21%, 17% and 26%, respectively. These findings are generally consistent with the INSTIs dolutegravir (DTG) and raltegravir (RAL) when coadministered with RBT. RBT increased DTG CL/F by 6% and reduced area under the concentration–time curve from time zero to 24 h (A U C0–24) and Cτ by 5% and 30%, respectively; Cmax increased by 16% [20]. Similarly, RBT increased RAL area under the concentration–time curve from time zero to 12 h (AUC0–12) by 19% and Cmax by 20% and decreased Cτ by 20% [21]. For both DTG and RAL, coadministration with RBT requires no dose adjustment [20,21]. However, neither the INSTI elvitegravir (EVG) nor the most recently approved INSTI, bictegravir (BIC), are recommended to be coadministered with RBT, as RBT reduced minimum observed plasma concentration (Cmin) by 67% and 56% for EVG and BIC, respectively, so as to avoid potential loss of therapeutic effect and development of resistance [22,23].
The observed modest reduction in CAB Cτ by RBT is not considered clinically significant as resulting CAB geometric mean Cτ was above exposures following CAB 10 mg once daily administration. This dose was shown to be safe and efficacious in combination with RPV in HIV-infected patients. In a Phase II dose-ranging study, oral CAB 10 mg once daily (with a backbone of two nucleoside reverse transcriptase inhibitors or RPV) maintained durable suppression of HIV infection in treatment-naive subjects with a geometric mean trough concentration of 1.35 mg/ml, which is below the geometric mean trough of 2.5 mg/ml observed following CAB 30 mg and RBT 300 mg once daily in this study. Similarly, CAB AUC0–τ of 81.7 μg*h/ml with concurrent RBT remained above that of the 10 mg oral dose of 45.7 μg*h/ml. Therefore, the moderate reduction in plasma CAB exposure by RBT observed in this study is not considered clinically relevant with respect to oral CAB. These data are expected to represent the worst-case scenario and suggest that interactions with CAB LA will be modest. Simulations will inform dosing strategies with RBT and CAB LA, and therefore coadministration of CAB LA with RBT is not currently recommended without further study. Importantly, an increase in RPV dose is required when given alongside RBT, which may further complicate coadministration of CAB-RPV with RBT.
Patients living with HIV and AIDS are at an increased risk of developing TB due to a compromised immune system. These two diseases cause a heavy health burden for developing countries, especially sub-Saharan Africa [24]. The risk of developing TB is approximately 16–27x greater in people living with HIV compared with uninfected individuals, and HIV coinfection is associated with 11% of TB cases globally. Sub-Saharan Africa has a disproportionately high amount of HIV-associated TB, accounting for approximately 86% of all deaths from TB–HIV coinfection in 2016 [25,26]. The guidelines for the treatment of HIV and TB recommend treating both diseases at the same time, rather than treating TB first before initiating HIV therapy [10,27]. ARV options are limited for patients taking antimycobacterial medications because of the significant DDIs that accompany the most commonly used drug, RIF. TB remains a very significant health risk to the HIV population and needs to be addressed. RBT is an alternative to RIF and has lower potential for interaction than RIF. These data support further studies evaluating the use of CAB with RBT as a potential treatment option in TB–HIV-coinfected patients as well as a possible treatment option for HIV-uninfected individuals receiving CAB monotherapy for PrEP, as CAB is likely to be used in areas highly endemic for both HIV infection and TB.
In conclusion, RBT may be administered with oral CAB without dose adjustment. A modest decrease in plasma CAB following CAB LA administration with RBT is expected, and simulations will be performed to inform potential dosing strategies with CAB LA. These study results support the potential coadministration of CAB + RBT in individuals requiring TB treatment.
Footnotes
Acknowledgements
Funding was provided by ViiV Healthcare UK Limited All authors contributed to the interpretation of the results, including critique of key findings, and drafting of the final version of the report for submission.
We would like to thank the clinical research staff, site investigator, and study volunteers for their participation in this study. We would also like to thank Joseph Piscitelli (University of North Carolina, School of Pharmacy) for his contributions to this manuscript. Professional medical writing and editorial assistance was provided by Sharmin Bovill at Articulate Science (Manchester, UK), funded by ViiV Healthcare.
This work was presented in part at the 19th International Workshop on Clinical Pharmacology of Antiviral Therapy, 22–24 May 2018, Baltimore, MD, USA.
SLF and NL are employees of GlaxoSmithKline; RD, WS and PP are employees of ViiV Healthcare; YL is an employee of PAREXEL International; and MK is an employee of the Addenbrooke's Hospital, Cambridge University Hospitals NHS Foundation Trust, seconded by 50% in the GSK's Clinical Unit Cambridge and has no relevant conflict of interest.