
Abstract
Background
Currently approved anti-HCV drugs, the direct-acting antivirals (DAAs), are highly effective and target the viral RNA replication stage of the HCV life cycle. Due to high mutation rate of HCV, drug resistant variants can arise during DAA monotherapy. Thus, a combination of DAAs is necessary to achieve a high response rate. Novel HCV inhibitors targeting the HCV late stage such as assembly and release may further improve combination therapy with the DAAs. Here we characterize one late stage-targeting candidate compound, 6-(4-chloro-3-methylphenoxy)-pyridin-3-amine (MLS000833705). Methods: We treated HCV-infected cells with MLS000833705 and other HCV inhibitors and examined HCV RNA and infectious titres. We evaluated the colocalization of HCV core and lipid droplets by confocal microscopy. We performed HCV core-proteinase K digestion assay and several lipid assays to study the mechanism of MLS000833705.
Results
We showed that MLS000833705 decreased extracellular HCV RNA levels more than intracellular HCV RNA levels in HCV infectious cell culture. Similarly, MLS000833705 reduced infectious HCV titres substantially more in the culture supernatant than intracellularly. Confocal microscopy showed that MLS000833705 did not affect the colocalization of HCV core protein with cellular lipid droplets where HCV assembles. HCV core-proteinase K digestion assay showed that MLS000833705 inhibited the envelopment of HCV capsid.
Conclusions
Our study demonstrates that MLS000833705 is a late-stage HCV inhibitor targeting HCV morphogenesis and maturation. Therefore, MLS000833705 can be used as a molecular probe to study HCV maturation and secretion and possibly guide development of a new class of HCV antivirals.
Introduction
HCV is a single-stranded positive-sense RNA virus that belongs to the Flaviviridae family. The genome size of HCV is approximately 9,600 bases and the genome encodes a single polyprotein of about 3,000 amino acids. The polyprotein is cleaved into 10 proteins by viral and host proteases. The structural proteins are encoded in the N-terminus of the HCV polyprotein and are designated as core, E1, E2 and p7. The non-structural (NS) proteins are involved in viral replication, assembly and release, and are designated as NS2, NS3, NS4A, NS4B, NS5A and NS5B [1,2].
HCV is a leading cause of chronic liver diseases throughout the world. Over 80 million people are infected chronically, which eventually develops into end-stage liver diseases such as cirrhosis and hepatocellular carcinoma [3]. Currently, there is no vaccine available. Direct-acting antivirals (DAAs) targeting HCV proteins greatly improve cure rate compared with the earlier therapy of pegylated interferon and ribavirin.
The first-generation DAAs, boceprevir and telaprevir, with pegylated interferon and ribavirin reached sustained virological response (SVR) of 68–75% for naive patients and to 59–88% for patients treated previously. However, these DAAs are indicated only in HCV genotype-1 patients [4,5]. Shortly after the first-generation DAAs, the second-generation DAAs were approved by the FDA. These second-generation DAAs have several improvements such as high cure rates for all HCV genotypes (about 90–95%) and interferon-free regimens. Second-generation DAAs target not only HCV NS3/4A protease, but also HCV proteins involved in replication such as NS5A and NS5B [6].
Although DAA regimens for treating HCV infection are highly effective, the error-prone nature of the RNA-dependent RNA polymerase may generate resistance-associated variants during viral replication, which eventually reduce susceptibility to the DAAs [7]. Thus, combination of multiple DAAs is necessary for effective treatment of HCV. Furthermore, even with combination DAA therapy, a small percentage of patients (5-10%) and more for difficult-to-treat patients (genotype-3, cirrhosis) fail treatment.
Future therapeutic development of HCV inhibitors would likely be focusing not only on HCV replication but also on other stages of HCV life cycle, such as entry, assembly and secretion. Among them, there are examples of HCV late-stage inhibitors. The grapefruit flavonoid naringenin [8], the core dimerization inhibitor SL209 [9], the imino sugars PBDNJ0804 [10] and deoxynojirimycin [11] have been shown to inhibit assembly and secretion.
Recently, our laboratory identified multiple candidate compounds as late-stage HCV inhibitors via high-throughput screen [12]. In the current study, we validated and characterized MLS000833705, a late-stage HCV inhibitor, as a molecular probe for viral assembly and potential candidate for therapeutic development.
Methods
Cell culture
Huh7.5.1 cells, Huh7 human hepatoma cell derived cell clones, were provided by Francis Chisari, The Scripps Research Institute, La Jolla, CA, USA [13]. Huh7.5.1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% (vol/vol) fetal bovine serum (FBS; Sigma-Aldrich, St Louis, MO, USA) and 1% penicillin Streptomycin (Corning, Corning, NY, USA) in 5% CO2 at 37°C.
Viral propagation and infection
HCV JFH1 strain (genotype-2a) was propagated and used for infection as previously described [13]. Full-length genomic JFH1 RNA was generated by in vitro transcription using MEGAscript T7 kit (Ambion, Foster City, CA, USA). For in vitro transcribed full-length genomic JFH1 RNA transfection, DMRIE-C reagent (Invitrogen) was used with 20 μg of JFH1 RNA and 2-3 million cells. The transfected cells were incubated in serum-free Opti-MEM (Invitrogen) for 6 h with occasional shaking before switching back to regular medium. Cells were then passaged every 2∼3 days and the virus containing medium was collected at the indicated time points.
For virus infections, one volume of the virus containing medium harvested from genomic JFH1 RNA transfected cells were used to inoculate naive Huh7.5.1 cells with additional four to nine volumes of fresh complete medium. The virus titre was measured by 50% tissue culture infectious dose (TCID50) assay and the concentration of virus containing medium was 1.2∼2.7x10 4 TCID50/ml.
Viral RNA isolation and quantification
Total RNA was extracted with GeneJET RNA Purification Kit (Thermo Fisher Scientific, Waltham, MA, USA) from cells, or with GeneJET Viral DNA and RNA Purification Kit (Thermo Fisher Scientific) from supernatants. Copy numbers of intracellular and extracellular HCV RNA were determined by quantitative RT-PCR with the probe and primers (Probe: 5′-/56-FAM/CTGCGGAACCGGTGAGTACAC/36-TAMSp/-3’, forward primer: 5′-CGGGAGAGCCAT-AGTGG-3’, reverse primer: 5′-AGTACCACAAGGC-CTTTCG-3’), using the Verso 1-step RT-qPCR Mix (low ROX; Thermo Fisher Scientific) on an ViiA 7 Real-Time PCR System (Thermo Fisher Scientific). The HCV RNA copy numbers were measured by standard curve which was generated with in vitro transcribed JFH1 RNA. PCR conditions are following. 1 cycle of 50°C for 2 min, 60°C for 30 min, and 95°C for 15 min, then followed by 50 cycles of PCR at 95°C for 20 s, and 62°C for 1 min.
Immunofluorescence
For HCV core protein staining, cells were fixed with 4% paraformaldehyde (ChemCruz, Dallas, TX, USA) 48-72 h after HCV infection. Then, the cells were permeabilized with 0.5% Triton X-100 (Tx-100, Sigma-Aldrich) in phosphate-buffered saline (PBS) and blocked by 3% bovine serum albumin (BSA; Sigma-Aldrich) in PBS. C1 anti-core antibody diluted in 0.1% Tx-100 in PBS with 1% BSA was used for staining HCV core protein. The cells were then incubated with Alexa Fluor 488 goat anti-mouse IgG (Invitrogen) or Alexa Fluor 555 donkey anti-mouse IgG (Invitrogen) in 0.1% Tx-100 in PBS with 1% BSA. For nuclei staining, 4’,6-diamidino-2-phenylindole dilactate (DAPI; Invitrogen) was used. After each step, the cells were washed with PBS twice. Images were captured by an AMG EVOS FL fluorescent microscope or Zeiss LSM 700 confocal microscope.
Proteolytic digestion protection assay
As previously described [14], the cells seeded in 6-well plates were collected 48 h after JFH1 infection into 170 μl of PK buffer (50 mM Tris-HCl [pH 8.0], 10 mM CaCl2, 1 mM dithiothreitol) and subjected to five cycles of freeze and thaw. Subsequently, 50 μl of lysates were then either treated or untreated with 1% Tx-100 for 1 h at room temperature and with 50 μg/ml of proteinase K (PK; Roche, Basel, Switzerland) for 1 h at 4°C. PK digestion was stopped by addition of 5 mM phenylmethylsulfonyl fluoride and followed by 10 min incubation at 4°C. The residual core was evaluated by western blot as previously described [15] and using anti-core antibody (C7-50; Thermo Fisher Scientific).
Lipid droplet staining and quantification of images In most cases, lipid droplet staining followed HCV core staining in order to see the colocalization of HCV core and lipid droplet. For lipid droplet staining, a stock of 1 mg/ml BODIPY 493/503 (Thermo Fisher Scientific) is added to the fixed cells at 1:1,000 dilution in PBS. After 30 min of incubation, the cells were washed with PBS twice and were followed by DAPI nuclei staining. Images were captured by Zeiss LSM 700 confocal microscope. HCV core and lipid droplet signal intensity were measured by ImageJ (National Institutes of Health, Bethesda, MD, USA). For HCV core and lipid droplet colocalization signal measurement, HCV core signal intensity was divided by colocalization signal intensity for normalization. For lipid droplet signal measurement only, lipid droplet signal intensity was divided by DAPI staining signal intensity for normalization.
TCID50 Assay and FFU Assay
Infectious HCV titre measurement was performed in 96-well clear bottom plates seeded with 1x10 4 Huh7.5.1 cells per well 1 day prior to infection. Viral supernatants were diluted serially by 10-fold in complete medium. After 48 h or 72 h of incubation, cells were immunostained for HCV core as mentioned above. For TCID50, it is counted as positive if a well has more than one HCV core-expressing cell [16]. For focus forming unit (FFU) assay, it is counted as positive if a well has more than one small discrete clusters which are HCV core-expressing cells [17].
Iodixanol density gradient assay
In order to differentiate HCV infectious particles from other viral particles, OptiPrep™ density gradient medium (60% [w/v] solution of iodixanol in sterile water; Sigma-Aldrich) was used. Iodixanol stock solution was mixed with HEPES buffer (40 mM HEPES, 270 mM NaCl, 10 mM KCl) to generate 5, 10, 25 or 50% of iodixanol. For 40% of iodixanol solution, HEPES was replaced with viral supernatant. Gradients were centrifuged in Optima L-100 XP Ultracentrifuge for 24 h at 50,000 rpm at 4°C. The fractions (250 ml each) were collected from the top of the gradient and were then tested HCV infectivity. The density of the fractions was determined by the absorbance at 340 nm and calculated by using a table provided by the manufacturer (OptiPrep™; Sigma-Aldrich).
Lipid assays
Huh7.5.1 cells were homogenized by a microtube homogenizer (Takara Biomasher Standard, Takara Bio, Kusatsu, Japan). The cell lysates were centrifuged and the supernatants were distributed in 96-well clear bottom assay plate. Then, triglyceride content was analysed by Triglyceride Quantification Kit (BioVision, Milpitas, CA, USA) according to the manufacturer's protocol. Total cholesterol and ApoB levels were quantified by Cholesterol/Cholesteryl Ester Assay Kit (Abcam, Cambridge, UK) and Human Apolipoprotein B ELISA Kit (Abcam) according to the manufacturer's protocol.
HCVsc, HCVpp, HCV Replicon and ATPlite Assay
Huh7.5.1 cells were seeded in white opaque 96-well plates and cultured overnight. For HCVsc and HCVpp assays, a single round defective HCV particle (HCVsc) and HCVpp-1a were used for infecting Huh7.5.1 cells, respectively. For HCV replicon assay, HCV replicon genotype-2a containing a luciferase reporter was transiently transfected into Huh7.5.1 cells. These cells were treated with compounds simultaneously. Cyclosporin A was used as a positive control. Luciferase signal was measured 48 h after the compound treatment using the Renilla luciferase assay system (Promega, Madison, WI, USA). In case of ATPlite assay, one day after infection, the infected cells were treated with increasing concentrations of tested compounds. Cytotoxicity in Huh7.5.1 by tested compounds was evaluated by the ATP-based cell viability assay with ATPlite assay kit (PerkinElmer, Waltham, MA, USA).
Statistical analysis
Data were represented as mean ± standard error of the mean. Experiments were performed in duplicates or triplicates. Statistical analysis was performed using GraphPad Prism software (GraphPad, San Diego, CA, USA). A P-value <0.05 was considered to be statistically significant.
Results
Discovery of a small-molecule HCV late-stage inhibitor
Previously, we performed a quantitative high-throughput screen of the Molecular Libraries Small Molecule Repository of about 350,000 chemicals to identify novel HCV inhibitors [12]. Using various cell-based HCV virological assays, we were able to assign the targeted stage(s) for most of these compounds in the HCV life cycle. As part of validation assays which were done previously, we did a two-part HCV core immunofluorescence staining assay, which allows us to distinguish between early and late stage of HCV infection [18]. Early stage inhibitors decrease HCV core-positive cells similarly in both parts, while late-stage inhibitors show higher inhibition in the second part than in the first part of the assay.
To explore novel HCV inhibitors targeting HCV late-stage life cycle, we selected one promising inhibitor, MLS000833705, for further characterization (Figure 1A). As shown in Table 1 and Figure 2, MLS000833705 has an efficacy of 96.9% and potency (50% effective concentration [EC50]) of 0.08 μM. To confirm that MLS000833705 targets the late stage of HCV life cycle, we tested it with HCV life cycle assays, including HCV entry with HCVpp assay, replication with HCV replicon assay, and early stage infection (HCV entry to replication) with HCVsc assay. Unlike cyclosporin A, which is a replication inhibitor, MLS000833705 had no effect on HCV entry and replication (Figure 1B–1D) [12,19].
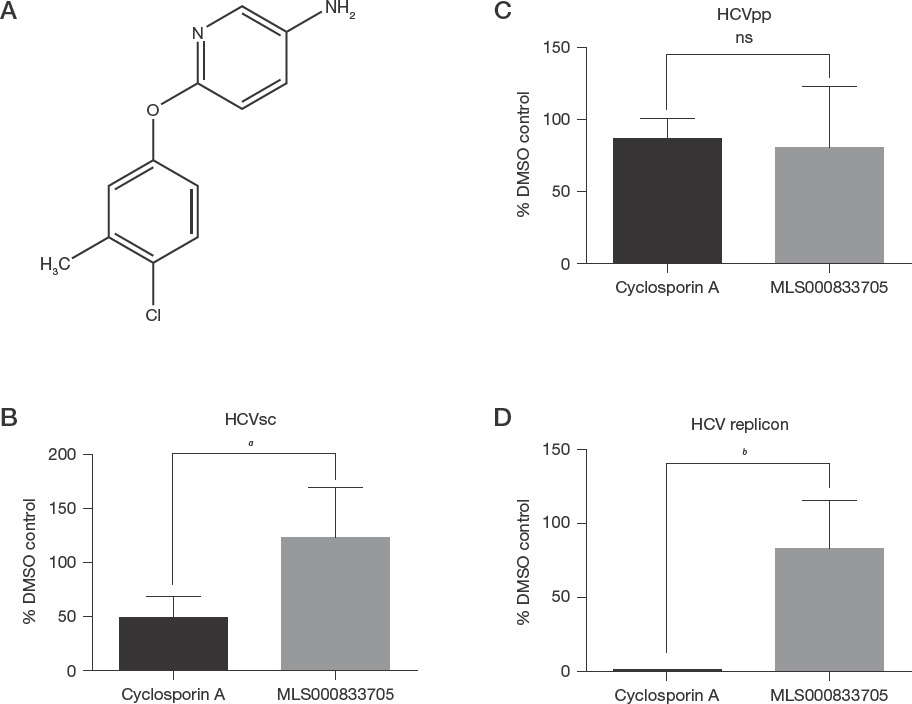
Characterization of MLS000833705 as an HCV late stage inhibitor
Chemical and anti-HCV properties of MLS000833705

EC50, 50% effective concentration.
MLS000833705 Differentially Inhibits Extracellular more than Intracellular HCV Levels
To evaluate the ability of MLS000833705 to inhibit HCV infection, we infected Huh7.5.1 cells with HCV strain JFH1 in the presence of MLS000833705 or cyclosporin A, which was used as a control. After 3 days, RNA was extracted from cells for intracellular and from medium for extracellular HCV RNA levels by RT-qPCR, respectively.
Cyclosporin A greatly decreased HCV RNA levels both intracellularly and extracellularly (Figure 2A). Compared with the mock-treated control, the highest drug concentration (10 μM) of cyclosporin A showed a ∼11,000-fold decrease of HCV RNA levels intracellularly and ∼8,000-fold decrease of HCV RNA levels extracellularly. Compared with the mock-treated control, the highest drug concentration (10 μM) of MLS000833705 exhibited a 1.5-fold decrease of HCV level intracellularly and 5.4-fold decrease of HCV level extracellularly. A two-part HCV core immunofluorescence staining assay had been used previously to distinguish between early and late-stage effects [19]. Early-stage inhibitors decrease HCV core-positive cells similarly in both parts, while late stage inhibitors show higher inhibition in the second part than in the first part of the assay. MLS000833705 exhibited more potent inhibition in the second part with EC50 of 0.5 μM than in the first part of the assay (EC50: 3.1 μM). This difference was also validated by direct measurement of the intracellular and extracellular HCV RNA levels (Figure 2B).
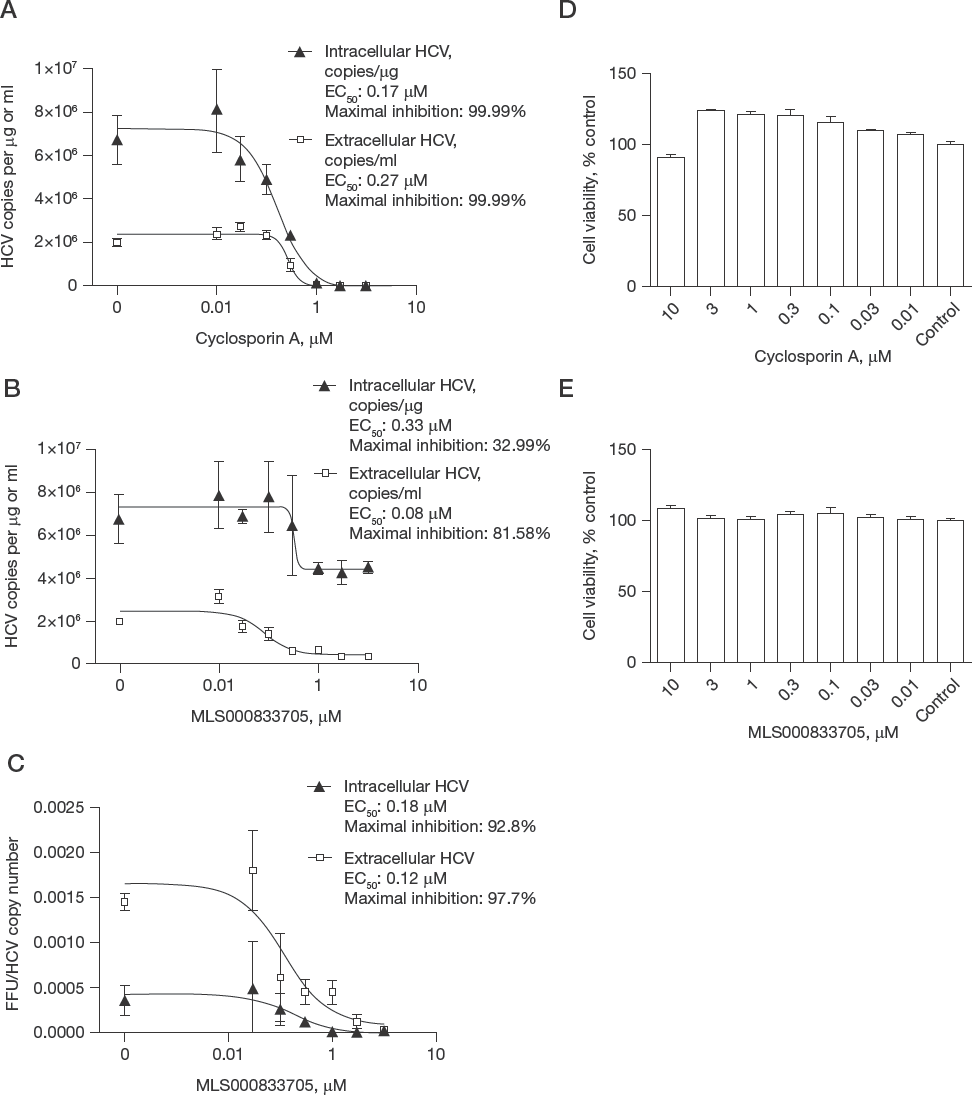
Characterization of MLS000833705 in the HCV life cycle Huh7.5.1 cells were infected with JFH1 in the presence of MLS000833705 or cyclosporin A at the indicated concentrations. The intracellular HCV RNA level and the extracellular HCV RNA level for MLS000833705 were compared with the intracellular HCV RNA level and the extracellular HCV RNA level from cyclosporin A 3 days after infection by RT-qPCR. Intracellular and extracellular HCV copy number of both (A) cyclosporin A and (B) MLS000833705 are drawn in one figure to show the fold difference between them. (C) Huh7.5.1 cells seeded in 6-well plates were infected with JFH1 and treated with MLS000833705 with indicated concentrations 6 h after JFH1 infection. 48 h later, intracellular and extracellular HCV particles were collected and re-infected naive cells in 96-well plates. 72 h later, the cells were fixed and stained with anti-core antibody and DAPI. Intracellular and extracellular focus forming unit (FFU) counts divided by HCV copy number are shown. (D&E) Huh7.5.1 cells were seeded in white opaque 96-well plates and cultured overnight. The next day, the cells were treated with increasing concentrations of cyclosporin A or MLS000833705. 48 h later, cytotoxicity in Huh7.5.1 by the compounds was evaluated by the ATP-based cell viability assay with ATPlite assay kit (PerkinElmer). Cell viabilities of (D) cyclosporin A treated cells and (E) MLS000833705 treated cells are shown, respectively.
To further determine the effects of MLS000833705 on intracellular and extracellular HCV infectious particles, we quantified the viral titres by focus-forming-unit assay and again showed a very dramatic effect on the extracellular (43-fold decrease) compared with the intracellular (14-fold decrease) viral levels (Figure 2C).
To exclude the possibility of a toxic effect of the compound, we conducted an ATPlite assay, which measures cell viability. Although cyclosporin A exhibited slight toxic effect at 10 μM, MLS000833705 did not show toxicity at any concentration tested from 0.01 to 10 μM (Figure 2D and 2E).
MLS000833705 does not Affect HCV Core Trafficking
HCV core protein localizes on lipid droplets and is an essential step for HCV assembly and/or release [20]. Many host factors are involved in the late stage of HCV life cycle including assembly and/or secretion [21]. The host factor diacylglycerol acyltransferase-1 (DGAT1) is known to interact with HCV core and is involved in the trafficking of core to lipid droplets [22]. In the presence of DGAT1 inhibitor, the trafficking of core protein is suppressed and viral assembly is thus impaired [22]. We examined whether MLS000833705 blocks HCV core trafficking and used DGAT1 inhibitor as a control. In the presence of DGAT1 inhibitor, core trafficking was suppressed and the colocalization between core and lipid droplets was reduced significantly (Figure 3A and 3B). On the contrary, microsomal triglyceride transfer protein (MTP) inhibitor, which is known to inhibit HCV lipidation and secretion [23,24], showed increased colocalization, likely because of the blockage of downstream events after core assembly on the lipid droplets. In the presence of MLS000833705, no obvious colocalization difference was observed (Figure 3A and 3B). Taken together, MLS000833705 does not inhibit core trafficking to lipid droplets and likely targets a step downstream of core trafficking, such as envelopment and/or secretion.

MLS000833705 has no effect on core trafficking and disrupts HCV core envelopment
MLS000833705 Inhibits Envelopment of HCV Capsid
We next examined the process of HCV capsid envelopment by measuring the resistance of intracellular core to PK digestion [14]. A full membrane-enveloped HCV core with lipid-laden envelope glycoproteins would protect the capsid protein, core, for protease digestion. Thus, the degree of envelopment can be quantified by the resistance of core to PK digestion. Cells were infected with HCV, lysed and treated with PK. Undigested core was quantified by western blot. Cell lysate pretreated with Tx-100 to solubilize all membranes before PK digestion were used as a control (shown as both PK and Tx-100-positive in the figure). Treatment with both PK and Tx-100 resulted in complete core proteolysis for both samples (Figure 3C). Treatment with PK alone resulted in a small fraction (∼7%) of core protected from PK digestion in control cells, while MLS000833705-treated sample showed almost complete disappearance of HCV core (Figure 3C), suggesting that the envelopment process was impaired.
MLS000833705 has no Effect on Triglyceride, Cholesterol, ApoB Protein and HCV Lipidation
HCV promotes its replication by regulating host cell lipid metabolism [25]. Since the late stage of HCV life cycle such as assembly and/or release is closely related to enhanced lipogenesis, we hypothesize that MLS000833705 may affect lipid metabolism resulting in suppression of the late stage of HCV life cycle [26,27]. To test our hypothesis, we measured triglyceride level in the presence of MLS000833705 in Huh7.5.1 cells. Total lipid was extracted from Huh7.5.1 cells, measured by a colorimetric assay, and normalized by protein content. Triglyceride level was increased in the cells treated with oleic acid (OA), which was used as a positive control (Figure 4A) [28]. There was no change in the cells treated with both MLS000833705 and DGAT1 inhibitor (Figure 4A and 4B).

MLS000833705 has no effect on triglyceride
To rule out the possibility that the basal level of triglyceride and lipid droplet formation in Huh7.5.1 cells may be too low to show a suppressive effect of MLS000833705, we stimulated lipid droplet formation by using varying concentrations of OA (Figure 4C). The lipid droplet levels were then assessed by using BODIPY 493/503 staining followed by confocal microscopy. The signal intensity was quantified by ImageJ (National Institutes of Health). As expected, OA increased the lipid droplets in a dose-dependent manner, and MLS000833705 had no additional effect on the lipid droplet formation (Figure 4D).
We then measured total cholesterol levels in MLS000833705-treated Huh7.5.1 cells. There was no significant change in cells treated with MLS000833705 (Figure 5A). We also tested the effect of MLS000833705 on ApoB protein level in the supernatant because HCV secretion is linked to ApoB secretion [23]. There was no significant change of ApoB in cells treated with MLS000833705 (Figure 5B).
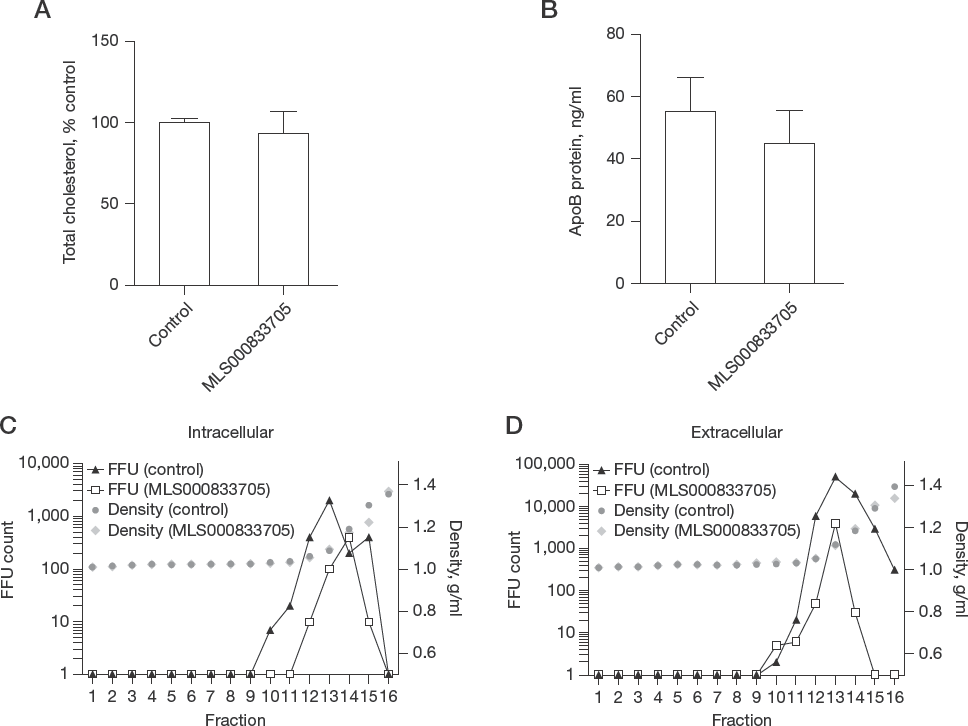
MLS000833705 has no effect on total cholesterol, ApoB protein and HCV lipidation
It has been shown that the densities of HCV particles are determined by their lipid content [29] and that lower lipoviroparticle density (higher lipidation) is associated with higher infectivity [30]. To define the role of MLS000833705 in the lipidation of HCV particles, we tested MLS000833705 or DMSO (control) with indicated concentrations in Huh7.5.1 cells. Both control and MLS000833705-treated groups showed comparable buoyant density for intracellular (Figure 5C) and extracellular HCV particles (Figure 5D), suggesting that MLS000833705 does not affect the lipid composition of HCV infectious particles.
Discussion
In recent years, treatment of HCV infection has improved significantly. As a result, HCV infection is now curable with the use of all-oral combinations of DAAs at a rate higher than 90% [6]. However, the high error-prone nature of the RNA-dependent RNA polymerase may produce resistance-associated variants during viral replication in the presence of DAA treatment. About 5–10% of patients still do not respond to current treatment, mainly because of emergence of drug-resistant variants [31]. To prevent drug resistance, combination DAA regimens are the gold standard for treating HCV to increase the barrier to resistance [32]. To further explore options for new combination regimen, we evaluated an HCV late-stage targeting compound, MLS000833705, in this study.
Various inhibitors have been reported to target other stages of HCV life cycle, such as entry [33–40]. Inhibitors targeting the late stage of HCV life cycle such as assembly or release have also been reported [8,10,26,41–43]. Moreover, some inhibitors can function in multiple stages of HCV life cycle. For example, iminodipyridinopyrimidine is shown to interfere with entry and release of HCV by targeting glycoprotein E1 in a genotype-specific manner [44]. Daclatasvir, in addition to inhibiting viral replication, can also efficiently block viral assembly [45]. In our previously reported high-throughput screen, we identified 47 candidate compounds as HCV late-stage specific inhibitors [12]. MLS000833705 appears to be the most potent and drug-like candidate.
In order to evaluate the HCV inhibitory activity and mechanism of action for MLS000833705, we performed various virological assays in the presence of MLS000833705. The data collectively support that MLS000833705 is involved in the late stage of the HCV life cycle. Since the late stage of HCV life cycle is closely related to lipid metabolism [46,47], we first tested whether MLS000833705 affects lipid metabolism. HCV particles assemble first on lipid droplets, which are the neutral lipids storage organelles in cells with triglyceride as the major component of lipid droplets [46,48]. We showed that unlike DGAT1 inhibitor, MLS000833705 does not affect the colocalization of HCV core with lipid droplets and thus is not involved in the trafficking of HCV core to the lipid droplet. Microsomal triglyceride transfer protein (MTP) has been shown to mediate the lipidation and envelopment of HCV particles in close association with lipid droplets after core assembly [23,49]. MTP inhibitor can block this step resulting in accumulation of HCV core particles in the lipid droplets [50]. Unlike MTP inhibitor, MLS000833705 did not cause accumulation of HCV core in the lipid droplets, suggesting that MLS000833705 acts on a step distinct from MTP in HCV morphogenesis. Finally, MLS000833705 had no effect on the triglyceride level and content of lipid droplets.
We next showed that HCV particles from the HCV core in MLS000833705-treated cells were more susceptible to PK digestion, suggesting a defect in envelopment. It is unlikely that MLS000833705 blocks envelopment completely because there was no significant accumulation of lipid droplet-associated core as shown above. The effect of MLS000833705 on HCV envelopment is consistent with the result that MLS000833705 inhibits the intracellular and extracellular infectious titres similarly (Figure 2C). MLS000833705 also does not seem to interfere with the viral secretion process per se, as there was a decrease in both intracellular and extracellular infectious HCV particles. It is possible that MLS000833705, by disturbing the envelopment process, may indirectly affect viral secretion. Together with the above data, we conclude that that MLS000833705 interferes with viral morphogenesis and maturation, that is, formation of mature infectious HCV virions with the envelope glycoproteins and accessory lipids.
Whether MLS000833705 directly targets the HCV glycoproteins or regulates a cellular process involved in the viral morphogenesis process is not yet clear. Future studies are necessary to further define the mechanism of action of MLS000833705 and its potential therapeutic development. This compound would also be a valuable molecular probe to elucidate the complex process of HCV maturation and secretion.
Footnotes
Acknowledgements
We thank Xin Hu (National Center for Advancing Translational Sciences, NIH, Bethesda, MD, USA) for his preparation of compounds, operating robot-controlled compound administration and plate reading. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
The authors declare no competing interests.