
Abstract
Background
The real-world effectiveness of pre-exposure prophylaxis (PrEP) may be influenced by circulating HIV strains resistant to either tenofovir or emtricitabine. Yet, few studies have examined rates of resistance to these drugs in clinical settings.
Methods
We conducted a retrospective cohort study of antiretroviral-naive participants in the Canadian Observational Cohort collaboration who initiated antiretroviral therapy between 2006 and 2014. In separate analyses, we determined the prevalence of pretherapy resistance and cumulative incidence of follow-up resistance to tenofovir and emtricitabine. We used multivariable proportional hazards models to examine associations between baseline variables and the development of resistance.
Results
We studied 6,622 antiretroviral-naive participants initiating therapy, of whom 5,428 (82.0%) had a baseline resistance test. Baseline resistance to tenofovir and emtricitabine was observed in 83 (1.5%) and 21 (0.4%) patients, respectively. Among patients without baseline resistance, the cumulative incidence of resistance to tenofovir and emtricitabine 5 years following treatment initiation was 0.0070 (95% CI 0.0046, 0.0095) and 0.033 (95% CI 0.028, 0.038), respectively. Following multivariable analysis, a baseline viral load ≥100,000 copies/ml was associated with emergence of tenofovir (hazard ratio [HR] 2.88; 95% CI 1.35, 6.15) and emtricitabine (HR 2.27; 95% CI 1.64, 3.15) resistance. Initiating an integrase inhibitor-based regimen and CD4+ T-cell count below 200 cells/mm3 were also associated with resistance to each drug.
Conclusions
We observed a low prevalence of baseline resistance and a low incidence of emergence of resistance to tenofovir and emtricitabine among antiretroviral-naive patients in routine clinical care.
Introduction
Pre-exposure prophylaxis (PrEP) with tenofovir disoproxil fumarate and emtricitabine has been shown to reduce the risk of HIV acquisition in populations at high risk for the disease, including men who have sex with men, transgender women, serodiscordant heterosexual couples and people who inject drugs [1–8]. Results from a meta-analysis of published trials and observational studies indicate that PrEP imparts a 51% reduction in the risk of HIV relative to placebo, a finding that was consistent across populations and modes of HIV acquisition [9]. Based on these data, the World Health Organization has recommended that PrEP be offered to all populations at risk of HIV [10]. Adherence has been identified as the most important determinant of PrEP effectiveness, with no protection against HIV demonstrated in trials where fewer than 40% of participants had detectable plasma levels of medication [9,11].
Another potentially important determinant of the effectiveness of PrEP is the prevalence of circulating HIV strains resistant to tenofovir and/or emtricitabine in the community of implementation. The prevalence of virus harbouring the K65R mutation is especially important, given that studies in macaques have correlated this mutation with PrEP failure, while no such effect was observed following exposure to strains containing the 184V mutation [12,13]. In this context, the protective activity of PrEP could be undermined, facilitating the transmission of drug resistant virus. Two recently published cases of infection with multi-drug resistant HIV in individuals with therapeutic levels of emtricitabine and tenofovir while receiving PrEP support this assertion and reinforces the need for ongoing population surveillance of tenofovir- and emtricitabine-resistant HIV [14,15]. Yet, few studies have characterized the incidence of resistance to these drugs in regions where PrEP is being introduced [16]. Consequently, surveillance for circulating strains of HIV resistant to tenofovir and/or emtricitabine has been identified as a research and policy priority as PrEP use increases [17,18]. These data are necessary to inform modelling of the effectiveness of PrEP and support regional demonstration projects designed to optimize its implementation. Accordingly, we examined the incidence of tenofovir- and emtricitabine-resistant HIV in a large cohort of antiretroviral-naive individuals living with HIV in Canada.
Methods
Study design and population
We conducted a retrospective study using data from the Canadian Observational Cohort (CANOC), an interprovincial collaboration of eight cohorts of antiretroviral-naive people with HIV aged 18 years and over who had initiated combination antiretroviral therapy after 1 January 2000 [19]. For this study, we included patients who initiated antiretroviral therapy between 2006 and 2014. We selected 2006 as the start date because it corresponds with the earliest Department of Health and Human Services recommendations for performing genotypic resistance testing on antiretroviral-naive patients and the widespread uptake of tenofovir-based antiretroviral regimens in Canada [20]. We excluded two sites with incomplete data on resistance testing. Data extraction was performed locally at the participating sites and pooled at the coordinating centre in Vancouver, BC. All participating cohorts have received ethical approval from their institutional boards to contribute data to CANOC.
Outcomes
The primary outcomes of the study were the occurrence of baseline and emergence of resistance to tenofovir or emtricitabine among antiretroviral-naive patients. We determined the proportion of patients with baseline resistance to either tenofovir or emtricitabine using all available genotypic resistance tests performed prior to the initiation of treatment. The cumulative incidence of resistance to either tenofovir or emtricitabine during follow-up was computed separately using Kaplan-Meier curves and excluding patients with baseline resistance to the corresponding drug. Patients with baseline resistance to both tenofovir and emtricitabine were not included in time-to-resistance analyses. In all analyses, the resistance interpretation was performed using the virco® TYPE HIV-1 system developed by Virco (now Janssen, Beerse, Belgium). This system makes use of a large database of linked HIV sequences, resistance phenotypes and clinical outcomes to predict a ‘virtual’ resistance phenotype from an input sequence [21,22]. For most drugs (including emtricitabine and tenofovir) the inferred resistance phenotypes are classified as susceptible, low-level or high-level resistance on the basis of clinical cutoffs derived from the analysis of linked clinical outcome data [23]. Accordingly, tenofovir resistance could be primarily defined by the presence of three or more thymidine analogue mutations or the presence of K65R or 69 insertion site mutations, and emtricitabine resistance primarily defined by the presence of M184V/I.
HIV drug Resistance Testing
The drug resistance testing programme in Quebec is performed on plasma-derived samples on all newly diagnosed patients, first genotyped with physician-designated clinical indication of primary infection (0–6 months after documented seroconversion) or chronic treatment-naive stage infection. Genotyping is also performed following antiretroviral treatment-failure (>100 copies/ml). Sequences (1467 bp) were generated using propriety Virco primers (Virco, BNBA, Mechelen, Belgium), spanning the entire protease gene and reverse transcriptase (RT) codons 1 to 400 [24]. Sequencing of select patients was also performed across the entire integrase gene as previously described [25]. Sequences were compared with HXB2 using Seqscape v2.5 (Applied Biosystems, Foster City, CA, USA) to identify minor or major resistance mutations.
Drug resistance testing for Ontario and Quebec was conducted in British Columbia. RNA extracted from plasma samples with plasma viral load >250 copies/ml underwent nested RT-PCR of HIV protease and reverse transcriptase, followed by standard Sanger sequencing on an ABI instrument. Automated sequence analysis was performed by the custom RECall software package with which nucleotide ‘mixtures’ are called when the secondary peak area exceeded >17.5% that of the primary peak in at least half of the reads [26]. The methodology has been previously described in more detail [27].
Statistical analysis
All analyses were performed using SAS software (version 9.4; SAS Institute Inc., Cary, NC, USA) and R 3.3.1 (R Development Core Team, Vienna, Austria). We conducted separate analyses of baseline resistance and the development of resistance during follow-up. For the first analysis, we examined the association between baseline resistance and clinical and demographic variables using multivariable logistic regression analysis among participants with baseline resistance testing. Next, we conducted separate time to event analyses using multivariable Cox proportional hazards regression to examine the associations between baseline clinical and demographic variables and the emergence of resistance to tenofovir or emtricitabine among those patients who did not have baseline resistance to one or both of these drugs. We adjusted all models for age, sex, ethnicity, HIV acquisition risk factor, baseline viral load and CD4+ T-cell count, calendar year of treatment initiation and Canadian province of residence. We also adjusted time-to-resistance models for type of antiretroviral regimen initiated (that is, protease inhibitor, non-nucleoside reverse transcriptase inhibitor, integrase inhibitor). To allow for a non-linear relationship between baseline CD4+ T-cell count and time to development of resistance, restricted cubic splines were incorporated into the proportional hazard models with knots at 30, 210, 340 and 680 cells/mm3. Missing values of covariates were imputed using a fully conditional specification method. A discriminant function was used to impute missing values of the categorical ethnicity variable and logistic regression models were used to impute missing values of binary variables ‘injection drug use’ and ‘men who have sex with men.’ All variables contained in the substantive model were used in the imputation models, as well as the binary outcome when imputing for logistic models and the event indicator and Nelson-Aalen estimator of the cumulative hazard when imputing for proportional hazards models [28].
Results
Study population
We studied 6,622 antiretroviral-naive patients initiating antiretroviral therapy during the study period. The majority (n=5,599; 84%) of participants were male and the median age was 40 years (IQR: 32–47; Table 1). The median duration of follow-up after treatment initiation was 3.8 years (IQR 1.9–6.0).
Baseline characteristics, overall and by receipt of baseline resistance test

Results are presented as n (%) or median (IQR). ACB, African, Caribbean and Black; ARV, antiretroviral; IDU, intravenous drug use; II, integrase inhibitor; MSM, men who have sex with men; NNRTI, non-nucleoside reverse transcriptase inhibitor; PI, protease inhibitor; VL, viral load.
Baseline resistance
Among 5,428 (82.0%) participants who had undergone baseline genotyping, resistance to tenofovir and emtricitabine was observed in 83 (1.5%) and 21 (0.4%) patients, respectively (see Additional files 1 and 2 for prevalence of individual mutations). Most tenofovir-associated resistance (n=80; 96%) was intermediate level (inferred 1.0- to 2.3-fold decrease in susceptibility). The K65R substitution was found in two (2.4%) isolates with tenofovir resistance, and seven (8.4%) cases were related to the accumulation of three or more thymidine analogue mutations. The majority (n=17; 81%) of emtricitabine resistance was related to the M184I/V mutation. Following multivariable analysis, individuals living in British Columbia (adjusted odds ratio [OR] 0.33; 95 CI 0.20, 0.54) and Quebec (OR 0.10; 95% CI 0.04, 0.29) were less likely to harbour tenofovir resistant virus at baseline relative to individuals living in Ontario (Table 2). Because of small numbers, we could not fit logistic regression models for baseline emtricitabine resistance.
Logistic regression model for presence of baseline tenofovir resistance
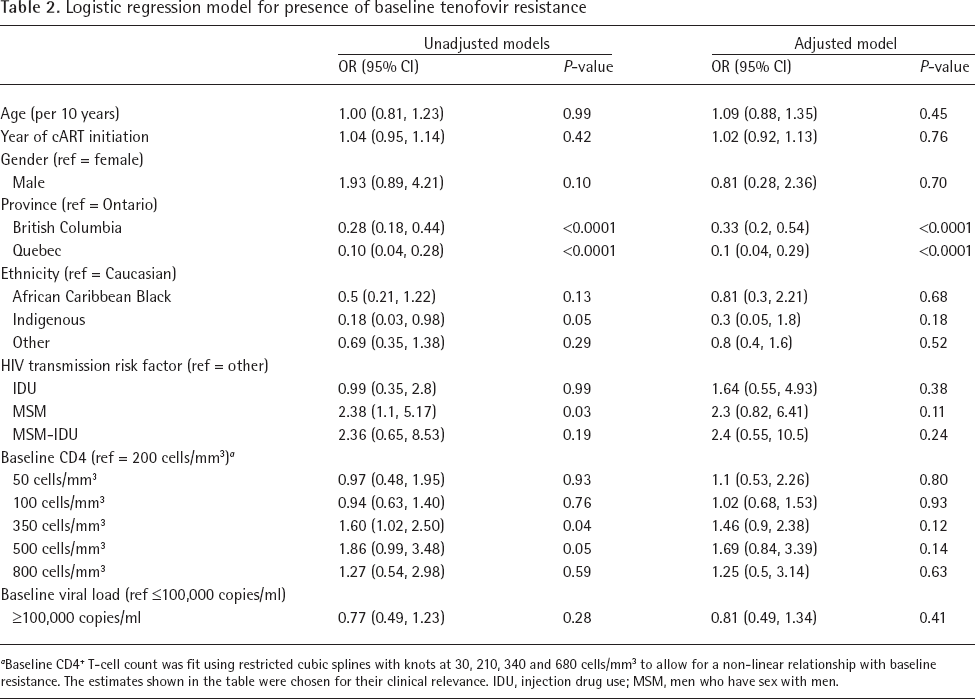
Baseline CD4+ T-cell count was fit using restricted cubic splines with knots at 30, 210, 340 and 680 cells/mm3 to allow for a non-linear relationship with baseline resistance. The estimates shown in the table were chosen for their clinical relevance. IDU, injection drug use; MSM, men who have sex with men.
Follow-up resistance
Among 6,539 patients with no baseline tenofovir resistance, the cumulative incidence of virological failure (viral load >50 copies/ml on two consecutive occasions at least 30 days apart or a viral load >1,000 copies/ml on a single occasion after initial suppression or 16 weeks on therapy) at 1, 3 and 5 years was 0.20 (95% CI 0.19, 0.21), 0.32 (95% CI 0.31, 0.33) and 0.38 (95% CI 0.36, 0.39). The corresponding cumulative incidences of having a resistance test were 0.15 (95% CI 0.14, 0.15), 0.23 (95% CI 0.22, 0.24) and 0.28 (95% CI 0.27, 0.29), respectively. After 1, 3 and 5 years following treatment initiation, the number of patients developing resistance to tenofovir was 16, 29 and 34, and the cumulative incidence of resistance to this drug was 0.0027 (95% CI 0.0014, 0.004), 0.0055 (95% CI 0.0035, 0.0075) and 0.0070 (95% CI 0.0046, 0.0095), respectively. For emtricitabine, the cumulative incidences of virological failure and resistance testing among 6,601 patients without baseline resistance to this drug were identical to those of tenofovir. Following treatment initiation, 74, 133 and 159 patients developed emtricitabine resistance at 1, 3 and 5 years, respectively, and the cumulative incidence of resistance at those time points were 0.012 (95% CI 0.0095, 0.015), 0.025 (95% CI 0.02, 0.029) and 0.033 (95% CI 0.028, 0.038). Tenofovir resistance was most often related to the K65R mutation (n=25; 69%) with the accumulation of multiple thymidine analogue mutations being observed infrequently (n=5; 14%). Resistance to emtricitabine was associated with the M184I/V mutations in nearly all (95%) cases (Additional files 1 and 2).
Following multivariable analysis, baseline viral load ≥100,000 copies/ml was associated with the emergence of resistance to tenofovir (hazard ratio [HR] 2.88; 95% CI 1.35, 6.15) and emtricitabine (HR 2.27; 95% CI 1.64, 3.15). Similarly, initiation of an integrase inhibitor-based regimen relative to a boosted protease inhibitor-based regimen was associated with emergence of resistance to tenofovir (HR 6.25; 95% CI 1.48, 26.29) and emtricitabine (HR 3.11; 95% CI 1.53, 6.35). In contrast, initiation of non-nucleoside reverse transcriptase-based (HR 2.97; 95% CI 1.33, 6.6) or unboosted protease inhibitor-based (HR 6.58; 95% CI 1.64, 26.32) therapy was associated with resistance to tenofovir, but not emtricitabine (Table 3). Demographic variables associated with emtricitabine resistance included Indigenous ethnicity (HR 3.79; 95% CI 2.27, 6.33) and African/Caribbean/Black (HR 2.65; 95% CI 1.38, 5.08) ethnicity (Table 3). In addition, relative to patients with a baseline CD4+ T-cell count of 200 cells/mm3, patients with lower CD4+ T-cell counts were more likely to develop resistance to either drug, whereas those with higher CD4+ T-cell counts were less likely to develop resistance (Figures 1 and 2). However, this relationship reached statistical significance only for emtricitabine (Table 2).
Adjusted proportional hazards models of times to tenofovir or emtricitabine resistance

Baseline CD4+ T-cell count was fit using restricted cubic splines with knots at 30, 210, 340 and 680 cells/mm3 to allow for a non-linear relationship with time to resistance. The estimates shown in the table were chosen for their clinical relevance. FTC, emtricitabine; HR, hazard ratio; IDU, injection drug use; II, integrase inhibitor; MSM, men who have sex with men; NNRTI, non-nucleoside reverse transcriptase inhibitor; PI, protease inhibitor; TDF, tenofovir disoproxil fumarate.

Adjusted hazard ratios of development of tenofovir resistance by CD4+ T-cell count
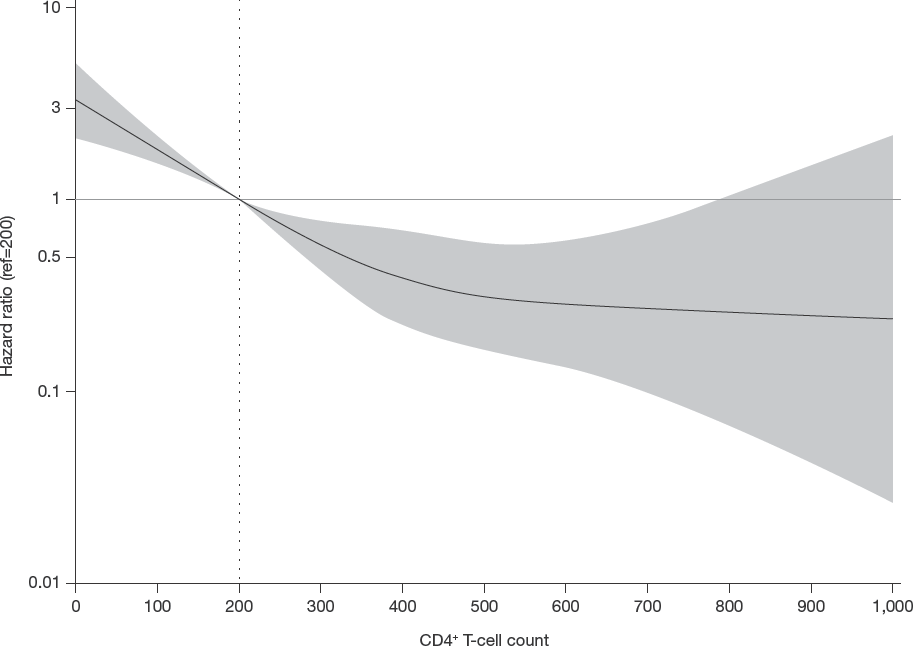
Adjusted hazard ratios of development of emtricitabine resistance by CD4+ T-cell count
Discussion
In our study of over 6,000 antiretroviral-naive individuals with HIV, we observed a low baseline and follow-up frequency of resistance to tenofovir and emtricitabine during a period of increasing use of these drugs. We found associations between resistance to these drugs and low baseline CD4+ T-cell count, high baseline viral load, Indigenous ethnicity and type of antiretroviral regimen initiated.
Several mechanisms likely explain our findings. The association between baseline resistance and province of residence is consistent with findings from the 2012/2013 Canadian HIV Strain and Drug Resistance Surveillance Program, which described the prevalence of drug resistance mutations in specimens obtained for laboratory confirmation of HIV [29]. This report demonstrated an overall higher burden of transmitted resistance in patients diagnosed in Ontario relative to British Columbia (14.7% versus 10.7%), with a greater proportion of specimens in Ontario being resistant to nucleoside analogues alone or with another class (that is, multidrug resistant). Because genotypes in our study are likely to represent samples obtained at a point in time following diagnosis but prior to treatment initiation, a low prevalence of baseline resistance would be expected as conditions would favour the selection of wild-type HIV strains rather than those harbouring nucleoside analogue mutations deleterious to viral fitness. However, this phenomenon should not vary by province, such that inter-provincial differences in the burden of baseline resistance should remain discernible.
The association between baseline viral load and emergence of drug resistance may reflect incomplete virological suppression following the initiation of treatment, and/or the presence of drug-resistant minority variants in CANOC participants living with HIV [30]. The association of emtricitabine resistance with high baseline viral load observed here and elsewhere likely reflects a biological association with reduced likelihood of achieving virological suppression [30]. A possible association between low baseline CD4+ T-cell count and antiretroviral resistance, particularly among nucleoside analogues, has been previously described [30,31]. Although a biological mechanism may exist for this finding, CANOC participants with HIV and low baseline CD4+ T-cell counts may differ from other patients in ways that could influence treatment outcomes, including social and structural determinants of access to HIV care [32,33]. In addition, findings from qualitative studies have demonstrated how social and structural determinants of health such as housing and food insecurity, institutionalized stigma, systemic racism and trauma can influence health-care access and produce healthcare disparities among Indigenous persons living in Canada [34–36]. Finally, the finding that patients initiating integrase inhibitor-based regimens were at greater risk for developing resistance may reflect the predominant use of raltegravir and elvitegravir during the study period, both of which were used as part of regimens which have a lower genetic barrier to resistance than ritonavir-enhanced protease inhibitor-based regimens. This assertion is supported by findings from comparative clinical trials demonstrating minimal resistance to supporting nucleoside backbones in patients experiencing virological failure with boosted protease inhibitor-containing regimens, whereas such resistance was common when virological failure occurred among patients randomized to either elvitegravir or raltegravir [37,38]. Since dolutegravir-based regimens impose a higher genetic barrier to resistance than elvitegravir and raltegravir [39], it is possible that our observed association between nucleoside resistance and integrase inhibitors would be attenuated with longer follow-up or data from individuals starting since it became available.
Our findings build upon those of earlier studies examining the frequency of resistance to tenofovir and emtricitabine among antiretroviral-naive patients. In one review of 203 studies describing 23,291 patients, the prevalence of mutations known to confer resistance to tenofovir was 0.4% [40]. However, most sequences available for analysis in this study were collected prior to 2004, a period preceding the widespread approval and availability of tenofovir. A more recent review that included 358 studies conducted in low- and middle-income countries and published between January 2012 and August 2017 found that the prevalence of tenofovir resistance was 3% among 56,044 genotypes reviewed [41]. However, this finding includes both antiretroviral-naive patients and pretreatment genotypes in those with a history of prior antiretroviral treatment. A study of 4,717 antiretroviral-naive patients seroconverting before January 2013 found that <1% of patients had high-level resistance to either tenofovir or emtricitabine [42], with similar results reported in a study of 1,318 patients diagnosed with primary HIV infection in France between 2007 and 2012 and 1,946 study participants with baseline resistance testing in the Strategic Timing of Antiretroviral Treatment (START) trial [43,44]. A more recent analysis encompassing the years 2005 to 2014 found the prevalence of tenofovir and emtricitabine mutations to be below 4% among antiretroviral-naive patients in Italy, with a slightly higher frequency of resistance to tenofovir among men who have sex with men (1.4%) relative to women (1.3%) and heterosexual men (0.0%) [16]. Similar findings were obtained from an analysis of 758 patients from the HIV Outpatient Study, in that the prevalence of the K65R and M184V mutations was 0.4% and 0.8%, respectively [45]. However, this finding likely underestimates the prevalence of tenofovir resistance because it does not consider the contribution of thymidine analogue mutations to the occurrence of this phenomenon. Our study addresses this limitation by using an algorithm for defining tenofovir resistance that also includes thymidine analogue mutations.
Our study is strengthened by the large sample size of antiretroviral-naive patients and the comprehensive ascertainment of resistance to tenofovir and emtricitabine from several clinical cohorts across Canada. However, our study has several limitations. First, we did not have data on the time between infection with HIV and baseline genotypic resistance testing; it is therefore possible that mutations conferring resistance to tenofovir or emtricitabine could have become archived in some individuals, underestimating the prevalence of resistance to one or both drugs. This is important when viewed in light of recent findings indicating that mutations associated with the component drugs of PrEP are no longer detected after 6 months of seroconversion [46]. Similarly, not all patients underwent genotyping prior to initiating therapy, thereby limiting our ability to accurately measure the frequency of drug resistance. However, these limitations are common to all clinical cohort studies undertaking surveillance for drug resistance. We fit non-parsimonious regression models to examine associations between demographic and clinical characteristics with the development of resistance. These findings should be interpreted cautiously and considered hypothesis generating given the small number of events, particularly when interpreting associations between resistance and regimen type. Finally, we are unable to directly address the impact of the level of resistance documented on the effectiveness of PrEP. Although our results provide a measure of reassurance that the reliability of PrEP is unlikely to be undermined by the frequency of resistant HIV observed in this analysis, further research is required to conclusively address this question and examine whether the impact of resistance varies among different sub-populations of persons with HIV.
In conclusion, we observed a low incidence of tenofovir and emtricitabine resistance among antiretroviral-naive patients in a large Canadian cohort of patients receiving routine care. As PrEP becomes increasingly used, ongoing surveillance of the frequency of drug resistance is required to inform delivery of this intervention and model its effectiveness among populations at high risk for HIV.
Footnotes
Acknowledgements
The authors would like to thank all of the CANOC participants for their valued information, Valerie Nicholson (BC Centre for Excellence in HIV/AIDS) for her review of the manuscript, Chanson Brumme and Conan Woods of the BC Centre for Excellence in HIV/AIDS for their assistance with assembling the mutation data and Bluma Brenner of Lady Davis Institute at McGill AIDS Centre for providing information about the assay for detecting drug mutations in Quebec.
The CANOC principal investigators consist of Centre investigator Robert Hogg and Centre site investigators, Curtis Cooper, Deborah Kelly, Marina Klein, Mona Loutfy, Nima Machouf, Julio Montaner, Stephen Sanche, Alexander Wong, Sharon Walmsley, Abigail Kroch, Rejean Thomas. CANOC co-investigators include Ann N Burchell, Tony Antoniou, Ahmed Bayoumi, Mark Hull, Bohdan Nosyk, Sean B Rourke, Chris Tsoukas, Angela Cescon, Michelle Cotterchio, Charlie Goldsmith, Silvia Guillemi, P Richard Harrigan, Marianne Harris, Sean Hosein, Sharon Johnston, Claire Kendall, Clare Liddy, Viviane Lima, David Moore, Alexis Palmer, Sophie Patterson, Peter Phillips, Nisha Andany, Hasina Samji, Marek Smieja.
CANOC is funded by the Canadian Institutes of Health Research (CIHR) through a Centres Grant (Centres for HIV/AIDS Population Health and Health Services Research [CIHR #02684]); 2 Operating Grants (HIV/AIDS Priority Announcement [CIHR #134047]; a Population and Public Health Grant [CIHR #136882]); a Foundation Grant (Expansion of Antiretroviral Therapy and its Impact on Vulnerable Populations in Canada and Global Settings [CIHR #143342]); and in collaboration with the CIHR Canadian HIV Trials Network [CTN #242]. AMB is supported by the Fondation Baxter & Alma Ricard Chair in Inner City Health at St. Michael's Hospital and the University of Toronto. TA is supported by a CIHR New Investigator and University of Toronto Department of Family and Community Medicine Clinician Investigator Award. MBK is supported by a Chercheur National Career Award from the Fonds de recherche du Québec - Santé (FRQ-S). JM is supported by the British Columbia Ministry of Health and by the US National Institutes of Health (R01DA036307). SW is supported through an Applied Health Research Chair from the OHTN.
The funders had no role in the design of the study, data collection, analysis, interpretation of data and the decision to publish the manuscript.
CC has served on advisory boards for AbbVie and Gilead Sciences. MBK reports grants from Merck and ViiV Healthcare and personal fees for consultancy from ViiV Healthcare, Bristol-Myers Squibb, and Merck. ML has served on advisory boards and spoken at CME events for ViiV Healthcare, AbbVie, Merck Canada Inc. and Gilead Sciences. JM is supported with grants paid to his institution by the British Columbia Ministry of Health and by the US National Institutes of Health (R01DA036307). He has also received limited unrestricted funding, paid to his institution, from AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck and ViiV Healthcare. JR is co-investigator on four projects outside the submitted work with in-kind contributions or financial support from Merck and Gilead Sciences. SW has served on advisory boards and spoken at CME events for ViiV, AbbVie, Merck, Gilead, Janssen and Bristol-Myers Squibb. RH reports personal fees from Gilead Sciences. For the remaining authors no conflicts of interest were declared.