
Abstract
Background
Cancer is a leading cause of death in HIV-infected patients in the era of combination antiretroviral therapy (cART). Yet, there are no specific guidelines for the combined use of cART and chemotherapy in HIV-infected cancer patients. The cellular enzyme thymidylate synthase (TS) catalyses the conversion of dUMP to TMP, which is converted to TDP and ultimately to TTP, a building block in DNA synthesis. TS inhibitors are recommended in some cancers, particularly non-small cell lung cancer (NSCLC). Because TS inhibitors modulate intracellular concentrations of endogenous 2′-deoxynucleotides, we hypothesized that TS inhibitors could impact the anti-HIV activity of nucleoside analogue reverse transcriptase inhibitors (NRTIs).
Methods
We evaluated gemcitabine and pemetrexed, two approved TS inhibitors, on the anti-HIV activities of NRTIs in infectivity assays using peripheral blood mononuclear cells (PBMCs) and in humanized mice.
Results
Gemcitabine enhanced the anti-HIV activities of tenofovir, abacavir and emtricitabine (FTC) in PBMCs. In contrast, pemetrexed had no effect on tenofovir, enhanced abacavir and, unexpectedly, decreased FTC and lamivudine (3TC) activities. Pemetrexed inhibitory effects on FTC and 3TC may be due to lower concentrations of active metabolites (FTCtp and 3TCtp) relative to their competing endogenous nucleotide (dCTP), as shown by decreases in FTCtp/dCTP ratios. Gemcitabine enhanced tenofovir while pemetrexed abrogated FTC antiviral activity in humanized mice.
Conclusions
Chemotherapy with TS inhibitors can have opposing effects on cART, potentially impacting control of HIV and thereby development of viral resistance and size of the reservoir in HIV-infected cancer patients. Combinations of cART and chemotherapy should be carefully selected.
Introduction
Cancer is a leading cause of death in patients with HIV infection in the current era of combination antiretroviral therapy (cART) [1]. However, there are currently no specific guidelines for the use of cART in HIV-infected cancer patients undergoing chemotherapy [2]. This absence reflects the paucity of data on treatments for these patients, who are less likely to receive cancer treatment and are often excluded from cancer clinical trials [3,4]. It also reflects that studies on HIV-infected cancer patients have generally focused on cancer aspects, leaving out details on antiretrovirals (ARVs) and HIV outcomes [5–8]. Available data on the combined use of cART and chemotherapy show conflicting results. Some studies recommend co-administration of cART and chemotherapy [9,10], whereas others recommend cART discontinuation [11–13]. Co-administration of cART during chemotherapy may help protect CD4+ T-cells from virus killing and also decrease the risk of HIV drug resistance [4,14,15]. However, combinations of cART and chemotherapy may increase drug toxicity [16]. While drug interactions have not been formally studied, an increased risk of side effects in cancer patients taking cART with chemotherapy versus cART alone and increased rates of virological failure among patients taking certain protease inhibitor-based cART or chemotherapy regimens have been observed [17,18], suggesting that clinically significant drug interactions do exist.
We set out to identify safe and effective combinations of ARVs and chemotherapy for HIV-infected cancer patients. We focused on treatments for HIV-infected patients with non-small cell lung cancer (NSCLC), which is the leading cause of cancer death in HIV infection [14,19]. Standard-of-care chemotherapy for NSCLC recommends treatments with pemetrexed (PTX) and gemcitabine (GCB), which suppress cell proliferation in part by inhibiting thymidylate synthase (TS) [20,21]. TS catalyses the conversion of dUMP to TMP, using 5,10-methylenetetrahydrofolate (5,10-CH2THF) as the methyl-group donor. Subsequently, TMP can be converted to TDP and then to TTP, which is required for DNA replication [22]. Previous studies have shown that targeting cellular enzymes from nucleotide biosynthesis pathways, such as ribonucleotide reductase and inosine monophosphate dehydrogenase, can enhance the anti-HIV activity of nucleoside analogue reverse transcriptase inhibitors (NRTIs) [23–26]. This antiviral enhancement is mediated by an increase in intracellular concentrations of NRTI active metabolite (ddNTP) relative to concentration of its endogenous competing nucleotide (dNTP). Higher ddNTP/dNTP ratios favour incorporation of ddNTP over dNTP in nascent HIV DNA during HIV reverse transcription, terminating viral synthesis. We hypothesized that inhibition of TS by GCB and PTX could impact the antiviral activities of NRTIs, with potential implications for HIV-infected patients with NSCLC. This hypothesis is based on the observation that TMP depletion by TS inhibitors leads to subsequent depletion of TTP, which, in turn, lowers dGTP concentrations through feedback mechanisms [20,21,27]. Depletion of TMP has variable effects on dATP and dCTP concentrations, depending on cell line type and on culture conditions [20,21,27]. In this study, we have evaluated the effects of PTX and GCB on the anti-HIV activities of available NRTIs in primary peripheral blood mononuclear cells (PBMCs) in vitro and in humanized mice.
Methods
Viruses and drugs
The HIV BaL strain was obtained from the NIH AIDS Repository (Germantown, MD, USA). Tenofovir (TFV), tenofovir disoproxil fumarate (TDF), emtricitabine (FTC), lamivudine (3TC), abacavir (ABC), zidovudine (AZT) and maraviroc (MVC) were obtained from the NIH AIDS Repository. Dolutegravir (DTG) was purchased from Toronto Research Chemicals (Toronto, ON, Canada). PTX, GCB and carboplatin (CPT) were purchased from Sigma (St Louis, MO, USA). Raltitrexed (RTX) was purchased from Apexbio (Houston, TX, USA).
Cells and infectivity assays
Human PBMCs were separated from buffy coats of HIV seronegative donors (New York Blood Center, NY, USA) by density centrifugation over Ficoll-Hypaque (Sigma). For infection, PBMCs were stimulated with 2.5 μg/ml phytohaemagglutinin (PHA; Roche, Indianapolis, IN, USA) for 3 days. Stimulated cells were infected by incubation with virus at a multiplicity of infection (MOI) of 0.001 for 2 h. PBMCs were then washed 3x with PBS and cultured in 5% CO2 at 37°C, in RPMI/10% FBS supplemented with 100 units/ml interleukin (IL)-2 (Roche) and drugs. PBMCs were seeded in 96-well flat-bottom plates at a density of 2x10 5 PBMCs/200 μl. Following 3 days of culture, half of the medium was replaced with fresh medium containing IL-2, chemotherapy drugs and ARVs. On day 7, HIV p24 antigen production in the culture supernatant was assayed by ELISA (Coulter, Hialeah, FL, USA). Cell proliferation was determined by a commercial MTT assay (Roche) or by counting viable cells following staining with Trypan blue.
Quantification of intracellular concentrations of dCTP and FTCtp in primary CD4+ T-cells
CD4+ T-cells were isolated from healthy donor PBMCs using immunomagnetic beads (Invitrogen). Quantification of concentrations of endogenous dCTP and FTC active metabolite FTCtp was done by a validated LC-MS/MS method [28], which has a calibration range of 0.02-20 ng/ml. Cells were lysed by incubation in cold 70:30 methanol: water for 15 min in ice and stored at −80°C until time of analysis. Following protein precipitation, the metabolites FTC and dCTP were extracted using internal standards 13 C5-TFVdp and 13 C9 15 N3-dCTP, respectively. Extracts were analysed by anion exchange chromatography on a Thermo BioBasic AX (50*2.1 mm, 5 μm) column before detection on an AB Sciex API-5000 triple quadrupole mass spectrometer. Precision and accuracy of this assay is within 20%.
Measurement of 2′-deoxycytidine kinase activity
We measured the effects of TS inhibitors on the enzymatic activity of 2′-deoxycytidine kinase (dCK) by using the PRECICE® dCK Phosphorylation Assay Kit (NovoCIB, Lyon, France). This is cell-free assay based on the competitive inhibition of 2′-deoxyinosine (dI) phosphorylation by dCK. In the absence of an exogenous competitor, dCK phosphorylates dI by using ATP as phosphate donor, forming dIMP. dIMP is then converted to dXMP by IMPDH, releasing NADH2. Levels of NADH2 are measured by absorbance at 340 nm. In the presence of an added competitor, such as FTC, the phosphorylation of dI, which is a poor substrate of dCK, is inhibited, resulting in decreased levels of NADH2.
Generation of humanized mice, quantification of plasma HIV RNA and lymphocyte subsets
Animal protocols were approved by the Institutional Animal Care and Use Committee, University of Maryland School of Medicine. NOD/scid-IL-2Rgc null (NSG) mice (5–7 weeks) were intraperitoneally (ip) injected with 10 7 PBMCs isolated from buffy coats of healthy donors. 3 weeks later, mice were screened for human lymphocytes in peripheral blood samples by flow cytometry analysis. Successfully reconstituted animals were ip injected with 15,000 units of 50% tissue culture infective dose (TCID50) of HIV BaL, followed by daily ip treatment with TS inhibitors and ARVs, alone and in combination. Drugs were used at the following doses: GCB (1 mg/kg/day), PTX (100 mg/kg), TDF (3 mg/kg) and FTC (60 mg/kg). These mouse doses are equivalent to human doses [29–31], except for TDF that was used at sub-inhibitory doses to detect potential antiviral changes in the presence of GCB. A control group was treated daily with PBS (ip). Animals were treated for 2 weeks and monitored daily for external signs of clinical deterioration. Blood samples were drawn from the retroorbital vein and analysed for plasma HIV RNA copy number by quantitative RT PCR using HIV gag primers and for human CD4/CD8 ratios (flow cytometry analysis), as described earlier [32].
Statistical analyses
50% effective concentration (EC50) values were determined by variable slope non-linear regression analysis. To test for significance in drug infectivity assays, anti-retroviral EC50 values were compared in the presence and absence of cancer drugs by Student's t-tests. Non-parametric Mann–Whitney tests were used to compare each treatment group and control group in animal studies. Statistical analyses were performed using GraphPad Prism (version 4.0). P≤0.05 was considered significant.
Results
Gemcitabine and pemetrexed have varying effects on the anti-HIV activities of NRTIs in PBMCs Prior to evaluating the effect of PTX and GCB, both of which modulate dNTP concentrations, on the antiviral activities of NRTIs, we quantified their impact on cell proliferation and HIV replication in the absence of ARVs in the PBMC infectivity assay (Figure 1). As control, we used CPT, which inhibits cell proliferation by affecting DNA repair mechanisms [33] but has no effect on dNTPs. For both PTX and CPT, inhibition of HIV p24 production paralleled inhibition of cell proliferation, suggesting that decreases in HIV production were due mainly to reductions in cell number. In contrast, GCB inhibited p24 production to a greater extent than it did cell proliferation, consistent with previous studies on the anti-HIV activity of GCB [31].
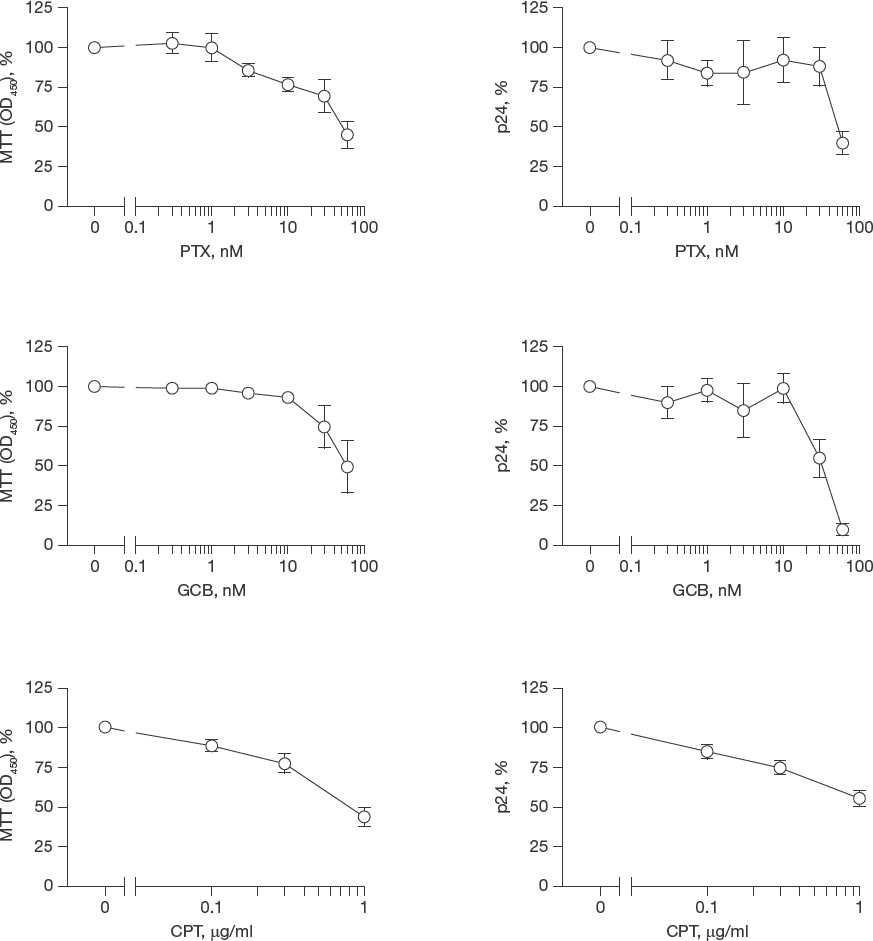
Effects of targeting TS with PTX and GCB on PBMC proliferation and HIV replication
We then evaluated the effects of PTX, GCB and CPT (control) on the potency of ABC (guanosine analogue), TFV (adenosine analogue), FTC (cytosine analogue) and AZT (thymidine analogue) in HIV-infected PBMCs as measured by p24 expression. Although AZT is rarely used in current cART, we included it in our studies to evaluate the impact of the cancer drugs on thymidine analogues. As an ARV control, we used MVC, a CCR5 blocker that inhibits HIV entry. Because of the antiproliferative effects of the cancer drugs in PBMCs, we performed our HIV experiments at 60 nM for PTX, 30 nM for GCB and at 1 μg/ml for CPT. These concentrations are within (albeit at the lower end) the range of reported EC50 values for cancer cells [34–36]. To account for decreases in PBMC numbers due to antiproliferation, we expressed the data as p24 per number of cells (p24 ng/10 6 cells). To evaluate changes in NRTIs potency, we compared EC50 values in the absence and presence of the cancer drug (Table 1). As expected, GCB, PTX and CPT did not significantly change the potency of CCR5 antagonist MVC. Also as expected, CPT did not significantly change the potency of any of the tested NRTIs. In contrast, GCB enhanced TFV, ABC and FTC potency by 6.8, 4.6 and 2.1-fold, respectively. GCB did not affect the potency of AZT. PTX enhanced ABC and AZT by 2.3 and 4-fold, respectively, but had no effect on TFV potency. However, PTX decreased FTC potency by 7.4-fold. The inhibitory effect of PTX on FTC was unexpected because PTX depletes dCTP in cell lines [20], and therefore we expected PTX to increase FTC activity. To confirm this unexpected negative impact of PTX on FTC, we next evaluated the effects of PTX on 3TC, another cytosine analogue NRTI. PTX suppressed HIV inhibition by 3TC (Additional file 1).
EC60 of TFV, ABC, FTC, AZT and MVC against HIV-1 BaL, in the absence and presence of cytotoxic drugs, in peripheral blood mononuclear cells

50% effective concentration (EC50) values are means ±SD from 3-5 experiments.
Fold reduction in EC50 compared with untreated cultures. (↓) indicates a significant decrease in EC50; (↑) indicates a significant increase in EC50.
P≤0.05 compared with untreated cultures by Student's t-test. ABC, abacavir; AZT, zidovudine; CPT, carboplatin; FTC, emtricitabine; GCB, gemcitabine; MVC, maraviroc; nd, not determined; PTX, pemetrexed; TFV, tenofovir.
PTX decreases FTCtp/dCTP ratios in primary CD4+ T-cells To gain initial insight into the mechanism by which PTX decreased the antiviral activity of FTC, we measured intracellular concentrations of dCTP and FTCtp and calculated FTCtp/dCTP ratios in primary CD4+ T-cells. Surprisingly, treatment with 60 nM PTX did not decrease dCTP concentrations as expected (Figure 2A). PTX treatment significantly decreased FTCtp/dCTP ratios (Figure 2B). The decreased FTCtp/dCTP ratio without a concomitant change on dCTP concentrations suggested that PTX could interfere with the synthesis of FTCtp. Therefore, we evaluated the impact of PTX on the enzymatic activity of dCK, which phosphorylates FTC to FTCmp, the first step in the phosphorylation cascade of FTC to FTCtp. Although the assay necessitated higher concentrations of FTC and PTX than those evaluated in our tissue culture experiments, the data showed that PTX does not inhibit dCK (Additional file 2). To understand whether PTX failure to decrease dCTP concentrations in primary cells might be related to feedback mechanisms resulting from inhibition of dihydrofolate reductase and glycinamide ribonucleotide formyltransferase, we evaluated RTX, a highly selective inhibitor of TS that does not inhibit other enzymes from nucleotide biosynthesis [22], in our culture system. RTX at 0.0T μM enhanced the activity of FTC by 5-fold, and those of ABC and TFV by 28- and 6-fold, respectively (Figure 3). These data support the idea that collateral inhibition of other enzymes by PTX decreases the antiviral activities of FTC and 3TC.
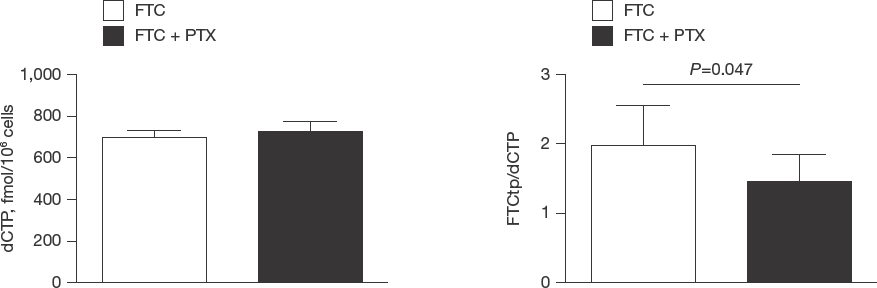
PTX does not decrease dCTP concentrations but decreases FTCtp/dCTP ratios in primary CD4+ T-cells

RTX, a highly selective TS inhibitor, enhances the antiviral activities of purine and pyrimidine NRTIs
Pemetrexed inhibition of emtricitabine reduces the antiviral potency of emtricitabine-containing cART
To test whether PTX could affect the antiviral potency of cART, we evaluated FTC/TFV/DTG activity in the presence and absence of PTX. FTC was used at its approximate EC90 against HIV BaL (30 nM), and TFV and DTG at their approximate EC50 values (3 μM for TFV and 0.T nM for DTG). As expected, PTX alone had little effect on HIV p24 levels. FTC/TFV/DTG suppressed p24 production below limits of detection (6 pg/ml). In contrast, FTC/TFV/DTG plus PTX failed to suppress p24 production (Additional file 3).
Gemcitabine enhances HIV inhibition by tenofovir in humanized mice
We next assessed the effects of TS inhibitors on the activities of NRTIs in vivo. We conducted these studies in NSG mice transplanted with human PBMCs (PBMC-NSG model). In a first experiment, we evaluated the effect of GCB (I mg/kg) on the anti-HIV activity of a sub-inhibitory dose (3 mg/kg) of TDF (prodrug of TFV) after TO daily ip injections. TDF was administered at a sub-inhibitory dose to detect potential antiviral changes in the presence of GCB. At the tested doses, monotherapy with either GCB or TDF decreased plasma HIV RNA slightly compared with PBS-treated controls, but differences were not statistically significant (Figure 4A). In contrast, GCB plus TDF decreased plasma HIV RNA by 6 log10 units compared with PBS controls (P=0.01 5). Consistent with the plasma viraemia data, mice treated with the GCB/TDF combination had significantly higher CD4/CD8 ratios compared with PBS control mice (P=0.025; Figure 4B). Animals treated with GCB and TDF, individually and in combination, did not have significant changes in body weight compared to controls, suggesting treatments were not overtly toxic (data not shown).
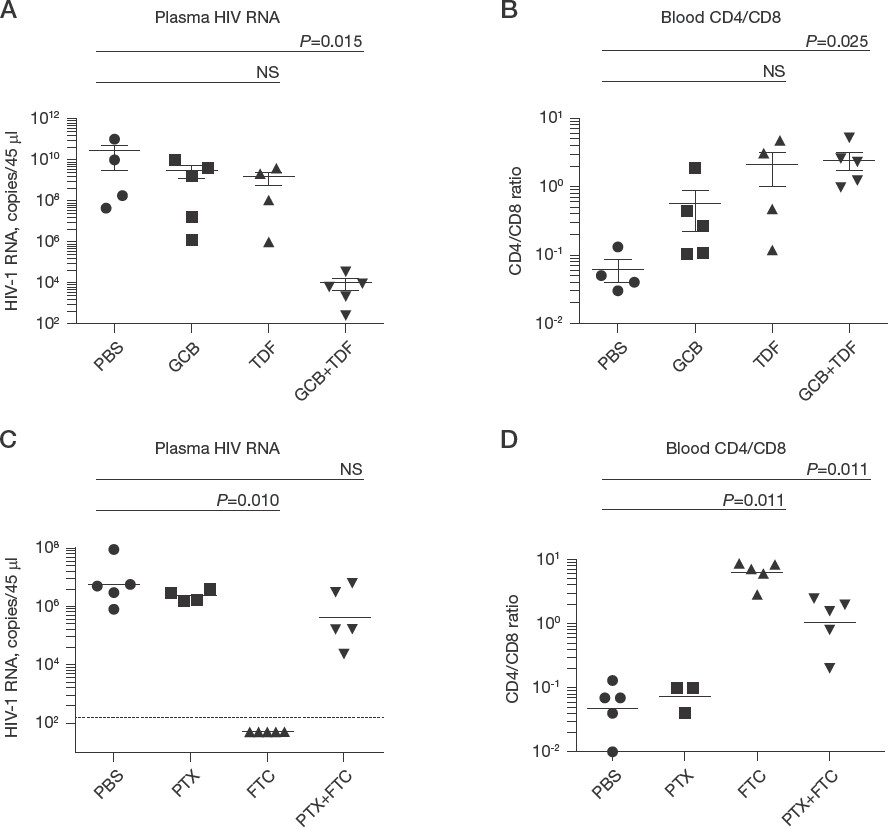
GCB enhances TDF and PTX inhibits FTC in humanized mice
Pemetrexed suppresses HIV inhibition by emtricitabine in humanized mice
We also evaluated the effect of PTX on the antiviral activity of FTC in the PBMC-NSG model. Infected mice were treated daily, via ip, with PTX (100 mg/kg) and FTC (60 mg/kg), alone and in combination. On day 10, as expected, PTX did not change plasma HIV RNA, whereas FTC decreased plasma HIV RNA to undetectable levels (P=0.010; Figure 4C). Plasma HIV RNA levels in PBS-control mice were lower than those in the previous experiment evaluating GCB and TDF; likely due to differences in PBMC susceptibility to HIV by the two donors used in these separate experiments. Importantly, in the PTX/FTC combination, PTX abrogated the effect of FTC on plasma HIV RNA. In agreement with the HIV RNA data, treatment with FTC alone gave the highest CD4/CD8 ratios, whereas ratios declined in the PTX/FTC combination (Figure 4D). Treatment with PTX and FTC, alone and in combination, were well tolerated by the mice, with no significant changes in body weight (data not shown).
Discussion
This study demonstrates that GCB and PTX, two TS inhibitors used in treatment of NSCLC and other cancers, have disparate effects on the anti-HIV activities of NRTIs in PBMC infectivity assays. GCB enhances TFV, ABC and FTC, but not AZT. In contrast, PTX enhances ABC and AZT, it has no effect on TFV, and decreases the activities of FTC and 3TC. We confirmed the enhancing effect of GCB on TFV and the inhibitory effect of PTX on FTC in humanized mice.
The data suggested that PTX and GCB modulate the antiviral activity of NRTIs by altering intracellular concentrations of endogenous dNTPs or their competing ddNTPs, thereby changing ddNTP/dNTP ratios. GCB enhancement of ABC, TFV and FTC activities is consistent with reduction of endogenous dGTP, dATP and dCTP pools via inhibitions of TS [21] and ribonucleotide reductase [37]. That GCB did not enhance AZT suggests that GCB treatment may restore the concentrations of TTP (endogenous competitor of AZTtp) via the salvage synthesis pathway, as shown previously with another inhibitor of ribonucleotide reductase [23]. PTX enhancement of ABC is consistent with PTX lowering dGTP pools (which is its anticancer mechanism [20]), and with primary lymphocytes relying almost exclusively on the de novo pathway for synthesis of dGTP [38]. That PTX did not substantially affect TFV antiviral activity is consistent with unchanged levels of dATP in PTX-treated cells [20]. However, PTX reduction of FTC and 3TC activities was unexpected because PTX depletes dCTP in cell lines [20]. We show that, contrary to data in cell lines, PTX does not decrease dCTP concentrations in primary CD4+ T-cells. PTX treatment decreased FTCtp/dCTP ratios, suggesting that PTX interferes with kinases mediating the phosphorylation of FTC to its active FTCtp metabolite. However, PTX did not inhibit dCK, which catalyses the phosphorylation of FTC to FTCmp, in an enzymatic assay. It is possible that PTX may interfere with other kinases downstream in the FTC phosphorylation pathway, such as 2′-deoxycytidine monophosphate kinase (dCMPK) and phosphoglycerate kinase 1 (PGK1) [39]. Experiments with RTX, a more selective TS inhibitor, suggested that the overall effect of PTX on dCTP concentrations might be the result of feedback mechanisms from secondary inhibition of dihydrofolate reductase and glycinamide ribonucleotide formyltransferase [40], two key enzymes in de novo synthesis of thymidine and purines.
There are, however, alternative potential mechanisms accounting for our results. One potential mechanism is that GCB may increase HIV mutation rates producing non-infectious virus, as shown previously in combinations of GCB and decitabine, a nucleoside analogue that incorporates in place of cytosine during minus-strand HIV DNA synthesis, but acts as guanosine because it pairs with cytosine [41]. However, this activity requires the combination of GCB with a nucleoside analogue that forms noncanonical base pairs. Because NRTIs do not form noncanonical base pairs, our data do not support this mechanism for the observed NRTI enhancement by GCB. Another potential mechanism relates to the different modes of action of GCB and PTX. GCB is a cytidine analogue whose triphosphate form (GCBtp) can compete with endogenous dCTP for incorporation into replicating cellular DNA (terminating its synthesis), whereas PTX does not incorporate into DNA. Thus, it is conceivable that GCBtp could interfere with HIV DNA synthesis by incorporation during reverse transcription. Yet, this activity does not appear to explain our data because GCB by itself, at concentrations that enhanced NRTIs, had only modest anti-HIV effects in vitro (Figure 1) or in vivo (Figure 4).
Our study has some limitations. The relatively low number of donors examined in our studies precludes evaluation of potential sexual differences in the effects of TS inhibitors on NRTIs. Although, to our knowledge, there is no evidence in the literature that TS inhibitors or the kinases phosphorylating NRTIs have different activities in males and females, we plan to assess the impact of gender in a future larger study. The experiments assessing the impact of PTX on HIV inhibition by the TFV/FTC/DTG combination demonstrated a negative impact of PTX when TFV and DTG were used at sub-inhibitory concentrations (at their EC50 values). We used sub-inhibitory concentrations of ARVs because higher concentrations completely suppressed virus production even in the presence of PTX, precluding assessment of PTX effects on ARV activities. Actual ARV concentrations in patients’ cells are above their EC90 values, implying that the inhibitory effects of PTX on FTC may be more limited in patients. Nevertheless, PTX suppression of human equivalent doses of FTC in the mouse model suggests that PTX effects on FTC may be relevant. Another limitation is that we evaluated the chemotherapy/ARV combinations in the PBMC-NSG model, in which duration of experiments is short because animals deteriorate with the development of graft versus host disease. However, immunodeficient mouse strains transplanted with PBMCs, such as the PBMC-NSG model, are valid models for testing drugs against HIV, as shown by previous studies [42–44]. A future study will evaluate these drug combinations in NSG mice transplanted with human CD34+ cells (HSC-NSG model), where mice can be monitored for months [32].
These data suggest that combinations of TS inhibitor-based chemotherapy and NRTIs should be carefully selected. Specifically, combinations of GCB with cART regimens containing TFV, ABC, FTC or 3TC are expected to have favourable anti-HIV interactions in cancer patients. Combinations of PTX and cART containing ABC or TFV are also expected to have favourable anti-HIV interactions, with those containing ABC giving the highest antiviral effect. These TS inhibitor/NRTI combinations, in conjunction with HIV integrase inhibitors, may be safely used in cancer patients undergoing chemotherapy because NRTIs and integrase inhibitors, unlike non-nucleoside reverse transcriptase inhibitors and protease inhibitors, have reduced potential for toxic interactions with chemotherapy [16]. These cART/chemotherapy combinations could circumvent the need for cART discontinuation during chemotherapy, ensuring HIV suppression and preventing HIV resistance. However, PTX reduction of FTC and 3TC antiviral activities is a concern. In the context of cART, PTX reduction of FTC or 3TC could translate into a 2-drug, rather than 3-drug, regimen, which might be insufficient to prevent emergence of drug resistance. Decreased cART potency could also fail to effectively suppress HIV replication in blood and tissues, favouring replenishment of infected cells in HIV reservoirs. Further studies evaluating the impact of PTX on FTC or 3TC-based combination ARV therapy are warranted. Together, these data could help delineate basic guidelines for the use of cART and chemotherapy with TS inhibitors in HIV-infected cancer patients. Because cancer responses to chemotherapy improve with effective control of HIV [45–49], we propose that optimal selection of cART/chemotherapy combinations could help control HIV and also to improve chemotherapy responses. This, in turn, could prolong survival of HIV-infected patients with NSCLC [50].
Footnotes
Acknowledgements
This work was supported by grants CFAR P30-AI-050410, CFAR 2P30-AI-050409, R01-CA-233441 and by additional research funds from the Institute of Human Virology.
The authors report no conflict of interest.